NUCYNTA ER Film-coated tablet Ref.[10904] Active ingredients: Tapentadol
Source: FDA, National Drug Code (US) Revision Year: 2019
12.1. Mechanism of Action
Tapentadol is a centrally-acting synthetic analgesic. The exact mechanism of action is unknown. Although the clinical relevance is unclear, preclinical studies have shown that tapentadol is a mu-opioid receptor (MOR) agonist and a norepinephrine reuptake inhibitor (NRI). Analgesia in animal models is derived from both of these properties.
12.2. Pharmacodynamics
Effects on the Central Nervous System (CNS)
Tapentadol produces respiratory depression, by direct action on the brainstem respiratory centers. The respiratory depression involves a reduction in the responsiveness of the brain stem respiratory centers to both increases in carbon dioxide tension and electrical stimulation.
Tapentadol causes miosis, even in total darkness. Pinpoint pupils are a sign of opioid overdose but are not pathognomonic (e.g., pontine lesions of hemorrhagic or ischemic origin may produce similar findings). Marked mydriasis rather than miosis may be seen with hypoxia in overdose situations.
Effects on the Gastrointestinal Tract and on Other Smooth Muscle
Tapentadol causes a reduction in motility and is associated with an increase in tone in the antrum of the stomach and duodenum. Digestion of food in the small intestine is delayed and propulsive contractions are decreased. Propulsive peristaltic waves in the colon are decreased, while tone is increased to the point of spasm, resulting in constipation. Other opioid induced effects may include a reduction in biliary and pancreatic secretions, spasm of the sphincter of Oddi, and transient elevations in serum amylase.
Effects on the Cardiovascular System
There was no effect of therapeutic and supratherapeutic doses of tapentadol on the QT interval. In a randomized, double-blind, placebo- and positive-controlled crossover study, healthy subjects were administered five consecutive immediate-release formulation doses of tapentadol 100 mg every 6 hours, tapentadol 150 mg every 6 hours, placebo and a single oral dose of moxifloxacin. Similarly, the immediate- release formulation tapentadol had no relevant effect on other ECG parameters (heart rate, PR interval, QRS duration, T-wave or U-wave morphology).
Tapentadol produces peripheral vasodilation which may result in orthostatic hypotension or syncope. Manifestations of histamine release and/or peripheral vasodilation may include pruritus, flushing, red eyes, sweating, and/or orthostatic hypotension.
Effects on the Endocrine System
Opioids inhibit the secretion of adrenocorticortropic hormone (ACTH), cortisol, and luteinizing hormone (LH) in humans [see Adverse Reactions (6.2)]. They also stimulate prolactin, growth hormone (GH) secretion, and pancreatic secretion of insulin and glucagon.
Chronic use of opioids may influence the hypothalamic-pituitary-gonadal axis, leading to androgen deficiency that may manifest as low libido, impotence, erectile dysfunction, amenorrhea, or infertility. The causal role of opioids in the clinical syndrome of hypogonadism is unknown because the various medical, physical, lifestyle, and psychological stressors that may influence gonadal hormone levels have not been adequately controlled for in studies conducted to date [see Adverse Reactions (6.2)].
Effects on the Immune System
Opioids have been shown to have a variety of effects on components of the immune system in in vitro and animal models. The clinical significance of these findings is unknown. Overall, the effects of opioids appear to be modestly immunosuppressive.
Concentration-Efficacy Relationships
The minimum effective plasma concentration will vary widely among patients, especially among patients who have been previously treated with potent agonist opioids. The minimum effective analgesic concentration of tapentadol for any individual patient may increase over time due to an increase in pain, development of a new pain syndrome, and/or potential development of analgesic tolerance [see Dosage and Administration (2.1, 2.3)].
Concentration-Adverse Experience Relationships
There is a relationship between increasing tapentadol plasma concentration and increasing frequency of dose-related adverse reactions such as nausea, vomiting, CNS effects, and respiratory depression. In opioid-tolerant patients, the situation may be altered by the development of tolerance to opioid-related adverse reactions [see Dosage and Administration (2.1, 2.2, 2.3)].
12.3. Pharmacokinetics
Absorption
The mean absolute bioavailability after single-dose administration (fasting) of NUCYNTA ER is approximately 32% due to extensive first-pass metabolism. Maximum serum concentrations of tapentadol are observed between 3 and 6 hours after administration of NUCYNTA ER. Dose proportional increases for AUC have been observed after administration of NUCYNTA ER over the therapeutic dose range.
Steady-state exposure of tapentadol is attained after the third dose (i.e., 24 hours after first twice daily multiple dose administration). Following dosing with 250 mg every 12 hours, minimal accumulation was observed.
Food Effect
The AUC and Cmax increased by 6% and 17%, respectively, when NUCYNTA ER tablet was administered after a high-fat, high-calorie breakfast. NUCYNTA ER may be given with or without food.
Distribution
Tapentadol is widely distributed throughout the body. Following intravenous administration, the volume of distribution (Vz) for tapentadol is 540 +/- 98 L. The plasma protein binding is low and amounts to approximately 20%.
Elimination
Metabolism
In humans, about 97% of the parent compound is metabolized. Tapentadol is mainly metabolized via Phase 2 pathways, and only a small amount is metabolized by Phase 1 oxidative pathways. The major pathway of tapentadol metabolism is conjugation with glucuronic acid to produce glucuronides. After oral administration approximately 70% (55% O-glucuronide and 15% sulfate of tapentadol) of the dose is excreted in urine in the conjugated form. A total of 3% of drug was excreted in urine as unchanged drug. Tapentadol is additionally metabolized to N-desmethyl tapentadol (13%) by CYP2C9 and CYP2C19 and to hydroxy tapentadol (2%) by CYP2D6, which are further metabolized by conjugation. Therefore, drug metabolism mediated by cytochrome P450 system is of less importance than phase 2 conjugation.
None of the metabolites contribute to the analgesic activity.
Excretion
Tapentadol and its metabolites are excreted almost exclusively (99%) via the kidneys. The terminal half-life is on average 5 hours after oral administration. The total clearance of tapentadol is 1603 +/-227 mL/min.
Specific Populations
Age: Geriatric Population
The mean exposure (AUC) to tapentadol was similar in elderly subjects compared to young adults, with a 16% lower mean Cmax observed in the elderly subject group compared to young adult subjects.
Hepatic Impairment
Administration of tapentadol resulted in higher exposures and serum levels to tapentadol in subjects with impaired hepatic function compared to subjects with normal hepatic function. The ratio of tapentadol pharmacokinetic parameters for the mild hepatic impairment group (Child-Pugh Score 5 to 6) and moderate hepatic impairment group (Child-Pugh Score 7 to 9) in comparison to the normal hepatic function group were 1.7 and 4.2, respectively, for AUC; 1.4 and 2.5, respectively, for Cmax; and 1.2 and 1.4, respectively, for t1/2. The rate of formation of tapentadol-O-glucuronide was lower in subjects with increased liver impairment.
Renal Impairment
AUC and Cmax of tapentadol were comparable in subjects with varying degrees of renal function (from normal to severely impaired). In contrast, increasing exposure (AUC) to tapentadol-O-glucuronide was observed with increasing degree of renal impairment. In subjects with mild (CLCR= 50 to <80 mL/min), moderate (CLCR= 30 to <50 mL/min), and severe (CLCR= <30 mL/min) renal impairment, the AUC of tapentadol-O-glucuronide was 1.5-, 2.5-, and 5.5-fold higher compared with normal renal function, respectively.
Drug Interaction Studies
Tapentadol is mainly metabolized by Phase 2 glucuronidation, a high capacity/low affinity system; therefore, clinically relevant interactions caused by Phase 2 metabolism are unlikely to occur. Naproxen and probenecid increased the AUC of tapentadol by 17% and 57%, respectively. These changes are not considered clinically relevant and no change in dose is required.
No changes in the pharmacokinetic parameters of tapentadol were observed when acetaminophen and acetylsalicylic acid were given concomitantly.
In vitro studies did not reveal any potential of tapentadol to either inhibit or induce cytochrome P450 enzymes. Only a minor amount of tapentadol is metabolized via the oxidative pathway. Thus, clinically relevant interactions mediated by the cytochrome P450 system are unlikely to occur.
The pharmacokinetics of tapentadol were not affected when gastric pH or gastrointestinal motility were increased by omeprazole and metoclopramide, respectively.
Plasma protein binding of tapentadol is low (approximately 20%). Therefore, the likelihood of pharmacokinetic drug-drug interactions by displacement from the protein binding site is low.
Alcohol
NUCYNTA ER may be expected to have additive effects when used in conjunction with alcohol, other opioids, or illicit drugs that cause central nervous system depression, because respiratory depression, hypotension, hypertension, and profound sedation, coma or death may result [see Warnings and Precautions (5.5)].
An in vivo study examined the effect of alcohol (240 mL of 40%) on the bioavailability of a single dose of 100 mg and 250 mg of NUCYNTA ER tablet in healthy, fasted volunteers. After co-administration of a 100 mg NUCYNTA ER tablet and alcohol, the mean Cmax value increased by 48% compared to control with a range of 0.99-fold to 4.38-fold. The mean tapentadol AUClast and AUCinf were increased by 17%; the Tmax and t½ were unchanged. After co-administration of a 250 mg NUCYNTA ER tablet and alcohol, the mean Cmax value increased by 28% compared to control with a range of 0.90-fold to 2.67-fold. The mean tapentadol AUClast and AUCinf were increased by 16%; the Tmax and t½ were unchanged.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
In mice, tapentadol HCl was administered by oral gavage at dosages of 50, 100 and 200 mg/kg/day for 2 years (up to 0.34 times in the male mice and 0.25 times in the female mice the plasma exposure at the maximum recommended human dose [MRHD] for NUCYNTA ER on an area under the time-curve [AUC] basis). No increase in tumor incidence was observed at any dose level.
In rats, tapentadol HCl was administered in diet at dosages of 10, 50, 125 and 250 mg/kg/day for two years (up to 0.20 times in the male rats and 0.75 times in the female rats the plasma exposure at the MRHD on an AUC basis). No increase in tumor incidence was observed at any dose level.
Mutagenesis
Tapentadol did not induce gene mutations in bacteria, but was clastogenic with metabolic activation in a chromosomal aberration test in V79 cells. The test was repeated and was negative in the presence and absence of metabolic activation. The one positive result for tapentadol was not confirmed in vivo in rats, using the two endpoints of chromosomal aberration and unscheduled DNA synthesis, when tested up to the maximum tolerated dose.
Impairment of Fertility
Tapentadol HCl was administered intravenously to male or female rats at dosages of 3, 6, or 12 mg/kg/day (representing exposures of up to approximately 0.56 times in the male rats and 0.50 times in the female rats the exposure at the MRHD on an AUC basis, based on extrapolation from toxicokinetic analyses in a separate 4-week intravenous study in rats). Tapentadol did not alter fertility at any dose level. Maternal toxicity and adverse effects on embryonic development, including decreased number of implantations, decreased numbers of live conceptuses, and increased pre- and post-implantation losses occurred at dosages ≥6 mg/kg/day.
13.2. Animal Toxicology and/or Pharmacology
In toxicological studies with tapentadol, the most common systemic effects of tapentadol were related to the mu-opioid receptor agonist and norepinephrine reuptake inhibition pharmacodynamic properties of the compound. Transient, dose-dependent and predominantly CNS-related findings were observed, including impaired respiratory function and convulsions, the latter occurring in the dog at plasma levels (Cmax), which are in the range associated with the maximum recommended human dose (MRHD).
14. Clinical Studies
14.1 Clinical Trials Summary
The efficacy of NUCYNTA ER was studied in five studies in patients with chronic pain and DPN. Efficacy was demonstrated in one randomized, double-blind, placebo- and active-controlled study in patients with chronic low back pain (LBP), and two randomized, double-blind, placebo-controlled studies in patients with pain related to diabetic peripheral neuropathy (DPN-1 and DPN-2).
14.2 Moderate to Severe Chronic Low Back Pain
In the LBP study, patients 18 years of age or older with chronic low back pain and a baseline pain score of ≥5 on an 11-point numerical rating scale (NRS), ranging from 0 to 10 were enrolled and randomized to 1 of 3 treatments: NUCYNTA ER, active-control (an extended-release Schedule II opioid analgesic), or placebo.
Patients randomized to NUCYNTA ER initiated therapy with a dose of 50 mg twice daily for three days. After three days, the dose was increased to 100 mg twice daily. Subsequent titration was allowed over a 3- week titration period to a dose up to 250 mg twice daily, followed by a 12-week maintenance period. There were 981 patients randomized. The mean age of the study population was 50 (range 18 to 89) years; the mean baseline pain intensity score was 8 (SD 1). Approximately half of the patients were opioid-naïve (had not taken opioids during the three months prior to the screening visit).
The number of patients completing the study was 51% in the placebo group, 54% in the NUCYNTA ER group and 43% in the active-control group. Lack of efficacy was the most common reason for discontinuation among placebo-treated patients (21%), whereas adverse events were the most common reason for discontinuation among the active treatment groups (17% and 32% for NUCYNTA ER and active-control, respectively).
After 15 weeks of treatment, patients taking NUCYNTA ER had a significantly greater pain reduction compared to placebo. The proportion of patients with various degrees of improvement is shown in Figure 1. The figure is cumulative, such that patients, whose change from baseline is, for example 50%, are also included at every level of improvement below 50%. Patients who did not complete the study were assigned 0% improvement.
Figure 1. Percentage of Patients Achieving Various Levels of Improvement in Pain Intensity - Study LBP1:
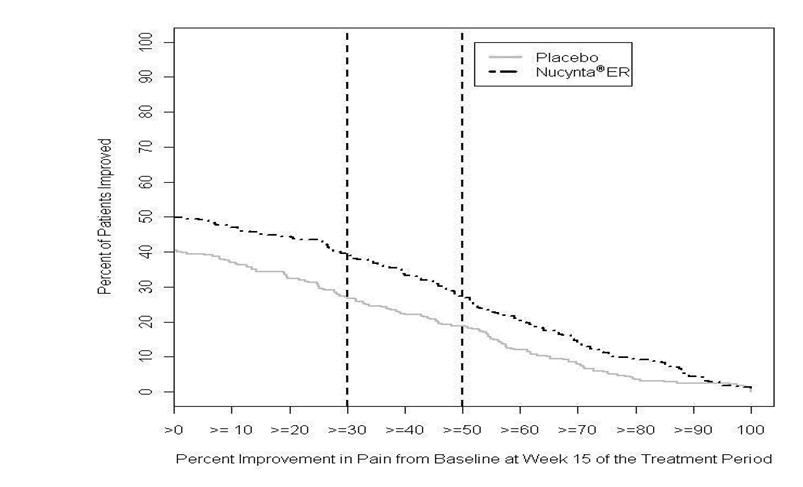
1 The last week of Study LBP was Week 15.
14.3 Neuropathic Pain Associated with Diabetic Peripheral Neuropathy
In the two DPN studies, patients 18 years of age or older with pain due to diabetic peripheral neuropathy and a pain score of ≥5 on an 11-point numerical rating scale (NRS) ranging from 0 (no pain) to 10 (worst possible pain) were enrolled. Following an open-label treatment period in which NUCYNTA ER was administered to all patients for three weeks and titrated to an individually stable dose, patients who had tolerated the drug and demonstrated at least a 1-point improvement in pain intensity on the NRS at the end of the open-label titration period were randomized to either continue the NUCYNTA ER dose (100 mg to 250 mg twice a day) reached during the open-label titration period, or receive placebo for 12 weeks of maintenance treatment. During the first 4 days of the double-blind maintenance period patients were permitted to take tapentadol ER 25 mg up to two times a day as additional medication. After the first 4 days, patients were allowed to take tapentadol ER 25 mg once daily as needed for pain, in addition to the patient's assigned study drug. Patients recorded their pain in a diary twice daily.
Study DPN-1
A total of 591 patients entered open-label treatment and 389 patients met the criteria for randomization into the double-blind treatment period. The mean age of the randomized population was 60 (range 29 to 87) years; approximately two-thirds of the patients were opioid-naïve (had not taken opioids during the three months prior to the screening visit).
During the titration period, 34% of patients discontinued open-label NUCYNTA ER. The most common reasons for discontinuation in the double-blind treatment period were lack of efficacy in the placebo group (14%) and adverse events in the NUCYNTA ER group (15%).
After 12 weeks of treatment, NUCYNTA ER provided a significantly greater reduction in pain intensity from baseline to the end of the 12-week double-blind period compared to placebo. Figure 2 displays the proportion of randomized patients achieving various degrees of improvement in pain intensity from the start of the open-label titration period to the last week of the randomized withdrawal period. The figure is cumulative, such that patients, whose change from baseline is, for example 50%, are also included at every level of improvement below 50%. Patients who did not complete the study were assigned 0% improvement.
Figure 2. Percentage of Patients Achieving Various Levels of Improvement in Pain Intensity - DPN-1:
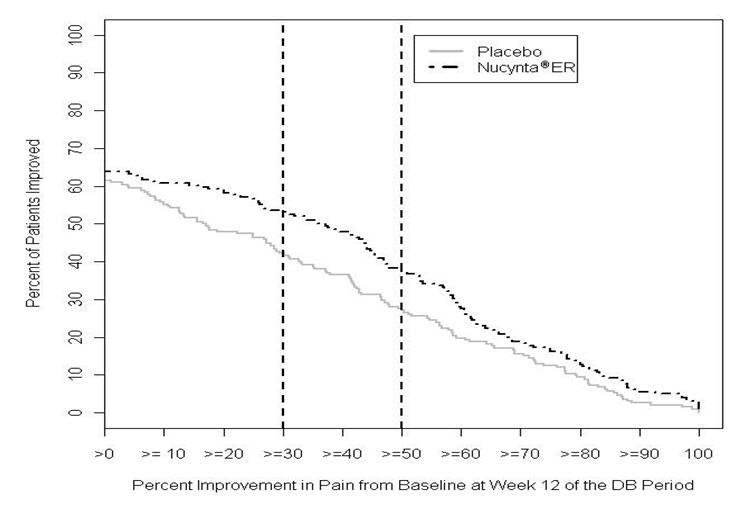
Study DPN-2
A total of 459 patients entered open-label treatment and 320 patients met the criteria for randomization into the double-blind treatment period. The mean age of the randomized population was 59 (range 28 to 83) years; approximately two-thirds of the patients were opioid-naïve (had not taken opioids during the three months prior to the screening visit).
During the titration period, 22% of patients discontinued open-label NUCYNTA ER and 6% of patients were not subsequently randomized because they failed to have at least 1-point improvement in pain intensity. The most common reason for discontinuation in the double-blind treatment period was adverse events in both the placebo group (9%) and the NUCYNTA ER group (14%).
After 12 weeks of treatment, NUCYNTA ER provided a significantly greater reduction in pain intensity from baseline to the end of the 12-week double-blind period compared to placebo. Figure 3 displays the proportion of randomized patients achieving various degrees of improvement in pain intensity from the start of the open-label titration period to the last week of the randomized withdrawal period. The figure is cumulative, such that patients, whose change from baseline is, for example 50%, are also included at every level of improvement below 50%. Patients who did not complete the study were assigned 0% improvement.
Figure 3. Percentage of Patients Achieving Various Levels of Improvement in Pain Intensity-DPN-2:
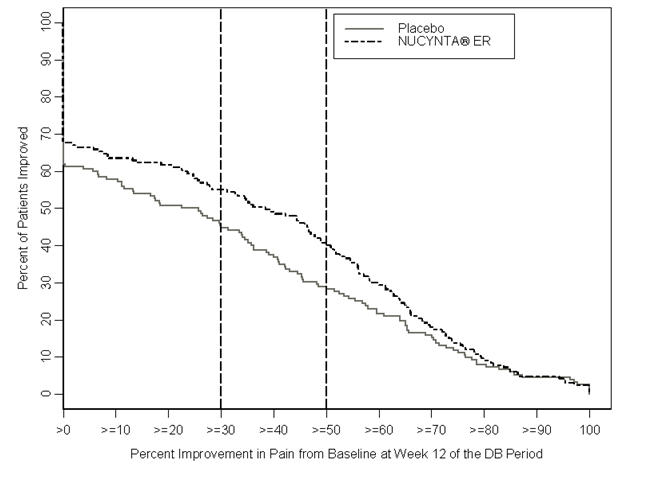
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.