OLINVYK Solution for injection Ref.[10350] Active ingredients:
Source: FDA, National Drug Code (US) Revision Year: 2020
12.1. Mechanism of Action
Oliceridine is a full opioid agonist and is relatively selective for the mu-opioid receptor. The principal therapeutic action of oliceridine is analgesia. Like all full opioid agonists, there is no ceiling effect to analgesia for oliceridine. Clinically, dosage is titrated to provide adequate analgesia and may be limited by adverse reactions, including respiratory, and CNS depression.
The precise mechanism of the analgesic action is unknown. However, specific CNS opioid receptors for endogenous compounds with opioid-like activity have been identified throughout the brain and spinal cord and are thought to play a role in the analgesic effects of this drug.
12.2. Pharmacodynamics
In nonclinical models, the antinociceptive effect of oliceridine can be antagonized by the opioid antagonist naloxone.
Effects on the Central Nervous System
Opioids produces respiratory depression by direct action on brain stem respiratory centers. The respiratory depression involves a reduction in the responsiveness of the brain stem respiratory centers to both increases in carbon dioxide tension and electrical stimulation.
Opioids causes miosis, even in total darkness. Pinpoint pupils are a sign of opioid overdose but are not pathognomonic (e.g., pontine lesions of hemorrhagic or ischemic origins may produce similar findings). Marked mydriasis rather than miosis may be seen due to hypoxia in overdose situations.
Effects on the Gastrointestinal Tract and Other Smooth Muscle
Opioids causes a reduction in motility associated with an increase in smooth muscle tone in the antrum of the stomach and duodenum. Digestion of food in the small intestine is delayed and propulsive contractions are decreased. Propulsive peristaltic waves in the colon are decreased, while tone may be increased to the point of spasm, resulting in constipation. Other opioid-induced effects may include a reduction in biliary and pancreatic secretions, spasm of sphincter of Oddi, and transient elevations in serum amylase.
Effects on the Cardiovascular System
Opioids produces peripheral vasodilation, which may result in orthostatic hypotension or syncope. Manifestations of histamine release and/or peripheral vasodilation may include pruritus, flushing, red eyes, sweating, and/or orthostatic hypotension.
Effects on the Endocrine System
Opioids inhibit the secretion of adrenocorticotropic hormone (ACTH), cortisol, and luteinizing hormone (LH) in humans [see Adverse Reactions (6.1)]. They also stimulate prolactin, growth hormone (GH) secretion, and pancreatic secretion of insulin and glucagon.
Chronic use of opioids may influence the hypothalamic-pituitary-gonadal axis, leading to androgen deficiency that may manifest as low libido, impotence, erectile dysfunction, amenorrhea, or infertility. The causal role of opioids in the clinical syndrome of hypogonadism is unknown because the various medical, physical, lifestyle, and psychological stressors that may influence gonadal hormone levels have not been adequately controlled for in studies conducted to date [see Adverse Reactions (6.1)].
Effects on the Immune System
Opioids have been shown to have a variety of effects on components of the immune system in in vitro and animal models. The clinical significance of these findings is unknown. Overall, the effects of opioids appear to be modestly immunosuppressive.
Concentration-Analgesia Relationships
In a fixed-dose bunionectomy trial (N=192), the onset of action of OLINVYK (as measured by the two-stopwatch method) was almost immediate (median 1-3 minutes) upon administration of the first dose. Perceptible pain relief was achieved in the majority of patients within 5 minutes after the first dose of OLINVYK.
The minimum effective analgesic concentration will vary widely among patients, especially among patients who have been previously treated with potent agonist opioids. The minimum effective analgesic concentration of oliceridine for any individual patient may increase over time due to an increase in pain, the development of a new pain syndrome, and/or the development of analgesic tolerance [see Dosage and Administration (2.1, 2.3].
Concentration-Adverse Experience Relationships
There is a general relationship between increasing oliceridine doses and increasing frequency of dose-related opioid adverse reactions such as nausea, vomiting, CNS effects, and respiratory depression. In opioid-tolerant patients, the situation may be altered by the development of tolerance to opioid-related adverse reactions [see Dosage and Administration (2.1, 2.2, 2.3)].
Cardiac Electrophysiology
The effect of oliceridine on the QTc interval was evaluated in 2 dedicated Thorough QT/QTc studies. A single-dose, randomized, positive- (moxifloxacin) and placebo-controlled 4 period crossover study evaluated the ECG effects of oliceridine at a therapeutic (3 mg IV infusion) and a supratherapeutic (6 mg IV infusion) dose in 62 healthy volunteers. Dose-dependent QTc prolongation (3 mg: 7 ms [upper 90% CI: 9 ms]; 6 mg: 12 ms [14 ms]), which occurred after peak oliceridine plasma concentration, was observed in this study.
A multi-dose, randomized, positive- (moxifloxacin) and placebo-controlled 3-way crossover study in 65 healthy volunteers evaluated intermittent dosing over 24 hours to the maximum daily cumulative dose of 27 mg. The maximum mean ΔΔQTcI was 11.7 ms (two-sided 90% UCI 14.7 ms) at 9 hours. Thereafter, the QTc effect did not progressively increase with repeat dosing, and despite continued dosing began to diminish after 12 hours.
The underlying mechanism and clinical significance of the transient QT changes seen in the single-dose and multiple-dose studies in healthy volunteers are unknown. These findings should be carefully considered when OLINVYK is administered in clinical settings where prolongation of the QT interval has been observed either due to the use of concomitant medications known to prolong the QT interval or to underlying medical conditions associated with QT interval prolongation.
12.3. Pharmacokinetics
Distribution
The mean steady-state volume of distribution of oliceridine ranges between 90-120 L indicating extensive tissue distribution. The plasma protein binding of oliceridine is 77%. In vitro data indicate that oliceridine is not an inhibitor of any of the major transporters, including breast cancer resistance protein (BCRP) and MDR1, at clinically relevant concentrations.
Elimination
Metabolism
In vitro studies suggest that oliceridine is metabolized primarily by CYP3A4 and CYP2D6 P450 hepatic enzymes, with minor contributions from CYP2C9 and CYP2C19 into inactive metabolites.
The mean clearance of oliceridine decreases slightly with increasing dose, resulting in greater-than-proportional exposure, particularly at doses greater than 2 mg. The percent of unchanged oliceridine excreted in the urine is low (0.97-6.75% of dose), reflecting its low renal clearance. The pharmacokinetics of oliceridine were not changed substantially (except for peak concentrations) when administered over different infusion times.
Excretion
Metabolic clearance is the major route of elimination of oliceridine, primarily by oxidation with subsequent glucuronidation. Additional biotransformation pathways included N−dealkylation, glucuronidation, and dehydrogenation. The majority of the metabolites (approximately 70%) are eliminated in the urine, with the remainder eliminated in the feces. Only a small amount of unchanged drug (0.97-6.75% of a dose) is found in the urine. The half-life of these metabolites (~44 hours) is much longer than that of unchanged oliceridine (1.3-3 hours). In vitro binding studies have demonstrated that none of these metabolites has any appreciable activity at the mu-opioid receptor.
Specific Populations
Renal Impairment
In a study comparing subjects with end stage renal disease (N=8) to healthy age and sex−matched healthy subjects (N=8), no significant difference in oliceridine clearance was observed. OLINVYK doses do not need to be adjusted in patients with renal impairment.
Hepatic Impairment
In a study of mild (N=8), moderate (N=8), or severe hepatic impairment (N=6), both clearance and total exposure were similar to age and sex-matched healthy controls (N=8). The mean half-life of oliceridine was increased in subjects with moderate (4.3 hours) or severe (5.8 hours) hepatic impairment, as compared with healthy subjects (2.1 hours), or patients with mild hepatic impairment (2.6 hours). The estimated volume of distribution of oliceridine was significantly higher in subjects with moderate or severe hepatic impairment (212 and 348 L, respectively), as compared to healthy subjects (126 L) or patients with mild hepatic impairment (167 L).
Based on these data, the initial dose of OLINVYK does not need to be reduced in patients with mild or moderate hepatic impairment, but these patients may require less frequent dosing. Use caution when dosing OLINVYK in patients with severe hepatic impairment. Consider reducing the initial dose, and administer subsequent doses only after a careful review of the patient's severity of pain and overall clinical status.
Drug Interaction Studies
In vitro studies suggest that oliceridine is metabolized primarily by the CYP3A4 and CYP2D6 P450 hepatic enzymes, with minor contributions from CYP2C9 and CYP2C19. Inhibition studies using selective inhibitors of all the major CYP enzymes show that only the inhibition of CYP3A4 and CYP2D6 significantly affects the metabolism of oliceridine in these assays, suggesting that the contribution of CYP2C9 and CYP2C19 to the metabolism of oliceridine is minor.
The effect of concomitant administration of a CYP2D6 inhibitor on the pharmacokinetics of OLINVYK, although not studied, may be similar to that noted in subjects who are CYP2D6 poor metabolizers. The plasma clearance of oliceridine in CYP2D6 poor metabolizers is approximately 50% of plasma clearance in subjects who are nonpoor CYP2D6 metabolizers [See Pharmacogenomics (12.5)].
In healthy subjects CYP2D6 poor metabolizers (n=4) given a single 0.25 mg dose of OLINVYK after 5 days of itraconazole 200 mg QD (a strong CYP3A4 inhibitor), the total exposure (AUC) of OLINVYK was increased by approximately 80%; however, the peak concentration was not significantly affected [See Pharmacogenomics 12.5]. The mean clearance of oliceridine was reduced to approximately 30% of that observed in nonpoor metabolizers of CYP2D6 [see Drug Interactions (7)].
Oliceridine does not inhibit any P450 enzymes at clinically relevant concentrations.
12.5. Pharmacogenomics
Oliceridine is metabolized by polymorphic enzyme CYP2D6. CYP2D6 poor metabolizers have little to no enzyme activity. Approximately 3 to 10% of Whites, 2 to 7% of African Americans, and <2% of Asians, generally lack the capacity to metabolize CYP2D6 substrates and are classified as poor metabolizers.
In healthy subjects who are CYP2D6 poor metabolizers, the AUC0−inf of oliceridine was approximately 2-fold higher than in subjects who are nonpoor CYP2D6 metabolizers [see Warnings and Precautions (5.6), Use in Specific Populations (8.8)].
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Long-term animal studies have not been completed to evaluate the carcinogenic potential of oliceridine.
Mutagenesis
Oliceridine was negative in an in vitro Ames bacterial reverse mutation assay, the in vitro chromosomal aberration assay using human peripheral blood lymphocytes, and the in vivo rat micronucleus assay.
Impairment of Fertility
In a fertility and early embryonic development study, oliceridine administered to female rats via continuous intravenous infusion at 6, 12, or 24 mg/kg/day for 14 days prior to cohabitation and through GD 15 for a total of 29-42 days resulted in prolonged estrous cycle lengths and decreased number of implantations and viable embryos at doses ≥12 mg/kg/day (≥3 times the estimated plasma exposure at the MRHD of 27 mg/day on an AUC basis).
Oliceridine did not alter male fertility at any dose tested. Males were dosed at 6, 12, or 24 mg/kg/day, producing plasma exposures up to 8 times the estimated plasma exposure at the MRHD, for 28 days prior to cohabitation, throughout the mating period and up to the time of scheduled necropsy for a total of 64-65 days of dosing.
13.2. Animal Toxicology and/or Pharmacology
Continuous intravenous infusion of oliceridine to rats for 14 days followed by one-day withdrawal from the treatment resulted in opioid withdrawal stress-related gastric lesions including erosions/ulcers in the glandular stomach, mucosal congestion/hemorrhage and degeneration/necrosis in the nonglandular stomach at all doses tested including the low dose producing plasma exposure 2 times the estimated human exposure at the MRHD on an AUC basis. The effect is believed to be due to acute withdrawal stress, as similar findings were not noted in rats sacrificed immediately after the last dose of oliceridine.
14. Clinical Studies
The efficacy of OLINVYK was established in two randomized, double-blind, placebo- and morphine-controlled studies of patients with moderate to severe acute pain following orthopedic surgery-bunionectomy or plastic surgery-abdominoplasty. In each study, pain intensity was measured using a patient-reported numeric rating scale (11-point numerical scale ranging from 0-10, where zero corresponds to no pain and 10 corresponds to worst pain imaginable).
In each study, patients were randomized to one of three OLINVYK treatment regimens, a placebo-control regimen, or a morphine-control regimen. Each blinded treatment regimen consisted of a loading dose, incremental doses delivered as needed via patient-controlled analgesia (PCA) device, and supplemental doses, beginning 1 hour after the initial dose, and hourly thereafter, as needed. The loading dose for all OLINVYK treatment regimens was 1.5 mg; demand doses were 0.1, 0.35 or 0.5 mg, according to the assigned treatment group; supplemental doses were 0.75 mg. The loading dose for the morphine treatment regimen was 4 mg; the demand dose was 1 mg; supplemental doses were 2 mg. The placebo-control regimen was volume-matched. A lockout interval of 6 minutes was used for all PCA regimens. For Studies 1 and 2, patients may have received rescue pain medication (pre-defined in the protocols as etodolac 200 mg every 6 hours, as needed) if the patient requested rescue pain medication and reported a Numeric Rating Scale score ≥4.
14.1 Study 1 – Orthopedic Surgery - Bunionectomy
A total of 389 patients (placebo n=79, OLINVYK 0.1 mg n=76, OLINVYK 0.35 mg n=79, OLINVYK 0.5 mg n=79, and morphine n=76), 19-74 years of age, with moderate to severe acute pain following orthopedic surgery-bunionectomy, were treated for up to 48 hours in Study 1 (NCT02815709). Treatment began after discontinuation of regional anesthesia in patients with pain intensity of ≥4 on a 0-10 numeric rating scale [NRS] within 9 hours after discontinuation of regional anesthesia. The analgesic effects were measured using the Summed Pain Intensity Differences over 48 hours (SPID-48). The SPID-48 is calculated by multiplying the Pain Intensity Difference (calculated by subtracting the pain intensity at a particular timepoint from the pain intensity at baseline) scores at each post-baseline timepoint by the duration (in hours) since the preceding timepoint, and then summing the values, over 48 hours. The majority of the study population was female (85%), and the mean age was 45 years. Patients were 69% White, 24% Black or African American, 4% Asian, 1% Native Hawaiian or Other Pacific Islander, 1% American Indian or Alaska Native and 1% other races. Twenty-five (25%) of patients were Hispanic or Latino.
The majority of patients treated with OLINVYK (0.1 mg treatment group: 83%; 0.35 mg treatment group 87%; 0.5 mg treatment group 84%) completed the randomized treatment period (compared to 60% of patients treated with placebo). Nine percent (9%), 4%, and 5% of patients in the 0.1 mg, 0.35 mg and 0.5 mg OLINVYK treatment groups, respectively, discontinued study medication due to lack of efficacy (compared to 34% of patients treated with placebo). In the 0.1 mg, 0.35 mg and 0.5 mg OLINVYK treatment groups, 41%, 20%, and 17% of patients, respectively, used the protocol-specified rescue medication etodolac, compared to 77% of patients treated with placebo.
The mean (SD) baseline pain intensity score was 6.7 (1.7). A statistically significantly greater analgesic effect was observed in both 0.35 mg and 0.5 mg OLINVYK treatment groups, compared to the placebo group (see Table 7). The mean pain intensities over time for placebo, 0.35 mg and 0.5 mg oliceridine and morphine treatment arms are shown in Figure 1.
Table 7. SPID-48 (Efficacy Endpoint) Results in Study 1 (Orthopedic Surgery - Bunionectom):
| Efficacy Measure | OLINVYK | |||
|---|---|---|---|---|
| Placebo regimen (N=79) | 0.35 mg regimen (N=79) | 0.5 mg regimen (N=79) | Morphine regimen (N=76) | |
| SPID-48 | ||||
| Average | 85 | 138 | 164 | 193 |
| Differencea | --- | 47.5 | 80 | 105 |
| 95% Confidence Interval | (19, 75) | (52, 108) | (77, 132) | |
a Treatments compared to placebo
Figure 1. Mean Pain Intensity versus Time Plot in Study 1:
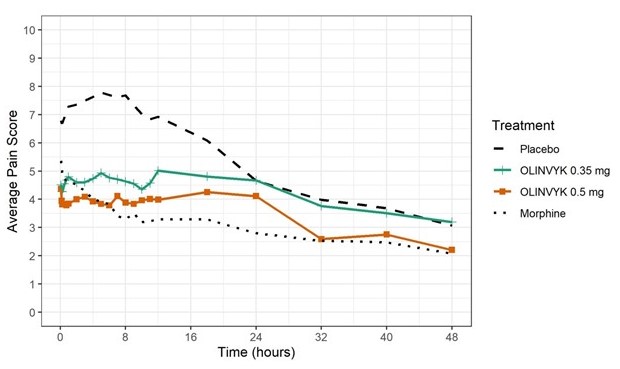
Note: Doses over the maximum recommended total cumulative daily dosage (27 mg) were treated as rescue medication in Figure 1. Pre-rescue pain scores were carried for 6 hours following the use of rescue medication.
In Study 1, 60% of patients in the OLINVYK 0.35 mg treatment group, and 63% of patients in the OLINVYK 0.5 mg treatment group reached the maximum recommended total cumulative daily dosage of 27 mg. The median (minimum) time to reach the 27 mg maximum recommended cumulative total daily dosage was 15.8 (9.1) hours for patients in the OLINVYK 0.35 mg treatment group, and 13.6 (6.8) hours for patients in the OLINVYK 0.5 mg treatment group.
14.2 Study 2 – Plastic Surgery - Abdominoplasty
A total of 401 patients (placebo n=81, OLINVYK 0.1 mg n=77, OLINVYK 0.35 mg n=80, OLINVYK 0.5 mg n=80, and morphine n=83), 20-71 years of age, with moderate to severe acute pain following plastic surgery-abdominoplasty, were treated for up to 24 hours in Study 2 (NCT02820324). The majority of the study population was female (99%), and the mean age was 41 years. Patients were 64% white, 31% Black or African American, 2% Asian, 1% Native Hawaiian or Other Pacific Islander, 0.2% American Indian or Alaska Native and 1% other races. Thirty-three (33%) of patients were Hispanic or Latino. Treatment began after discontinuation of general anesthesia in patients with NRS ≥5 within 4 hours after end of surgery. The analgesic effects were measured using the Summed Pain Intensity Differences over 24 hours (SPID-24).
The majority of patients treated with OLINVYK (0.1 mg treatment group: 86%; 0.35 mg treatment group: 90%; 0.5 mg treatment group: 87%) completed the randomized treatment period without discontinuing study medication (compared to 74% of patients treated with placebo). Eleven percent (11%), 3%, and 5% of patients in the OLINVYK 0.1 mg, 0.35 mg and 0.5 mg treatment groups, respectively, discontinued study medication due to lack of efficacy (compared to 22% of patients treated with placebo). In the OLINVYK 0.1 mg, 0.35 mg, and 0.5 mg treatment groups, 31%, 21%, and 18% of patients, respectively, used protocol-specified rescue medication etodolac, compared to 49% in patients treated with placebo.
The mean (SD) baseline pain intensity score was 7.3 (1.5). A statistically significantly greater analgesic effect was observed in the OLINVYK 0.5 mg and 0.35 mg treatment groups, compared to the placebo group (see Table 8). The analgesic effect was not significantly better in the OLINVYK 0.1 mg treatment group than in the placebo group. The mean pain intensities over time for placebo, 0.35 mg and 0.5 mg oliceridine and morphine treatment arms are shown in Figure 2.Table 8. SPID-24 (Efficacy Endpoint) Results in Study 2 (Plastic Surgery-Abdominoplasty Study):
| Efficacy Measure | OLINVYK | |||
|---|---|---|---|---|
| Placebo regimen (N=81) | 0.35 mg regimen (N=80) | 0.5 mg regimen (N=80) | Morphine regimen (N=83) | |
| SPID-24 | ||||
| Average | 75 | 90 | 94 | 103 |
| Differencea | --- | 14 | 18 | 30 |
| 95% Confidence Interval | (2, 26) | (5, 30) | (17, 42) | |
a Treatments compared to placebo
Figure 2. Mean Pain Intensity versus Time Plot in Study 2:
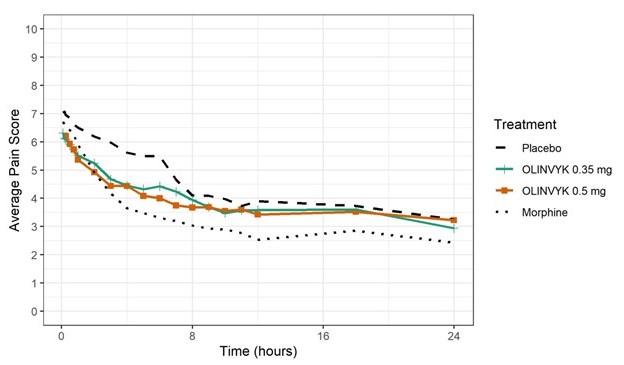
Note: Doses over the maximum recommended total cumulative daily dosage (27 mg) were treated as rescue medication in Figure 2. Pre rescue pain scores were carried for 6 hours following use of rescue medication. In Study 2, 28% of patients in the 0.35 mg dose group, and 43% of patients in the 0.5 mg dose group, reached the maximum recommended 27 mg total cumulative daily dosage. The median (minimum) time to reach the 27 mg maximum recommended cumulative total daily dosage was 19.4 (8.3) hours for patients in the 0.35 mg dose group, and 14.1 (6.4) hours for patients in the 0.5 mg dose group.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.