ONCASPAR Solution for injection/infusion Ref.[9116] Active ingredients: Pegaspargase
Source: European Medicines Agency (EU) Revision Year: 2020 Publisher: Les Laboratoires Servier, 50, rue Carnot, 92284, Suresnes cedex, France
Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic and immunomodulating agents, other antineoplastic agents
ATC code: L01XX24
Mechanism of action
The mechanism of action of L-asparaginase is the enzymatic cleavage of the amino acid L-asparagine into aspartic acid and ammonia. Depletion of L-asparagine in blood results in inhibition of protein-synthesis, DNA-synthesis and RNA-synthesis, especially in leukaemic blasts which are not able to synthesise L-asparagine, thus undergoing apoptosis.
Normal cells, in contrast, are capable of synthesising L-asparagine and are less affected by its rapid depletion during treatment with the enzyme L-asparaginase. The PEGylation does not change the enzymatic properties of L-asparaginase, but it influences the pharmacokinetics and immunogenicity of the enzyme.
Pharmacodynamic effects
Anti-leukaemic effect of L-asparaginase is related to a sustained L-asparagine depletion in blood and cerebrospinal fluid (CSF). The pharmacodynamic (PD) effect of Oncaspar was assessed after IM (Study CCG-1962) and IV administration (AALL07P4).
In Study CCG-1962, PD effect of Oncaspar was assessed through serial measurements of asparagine in serum (n=57) and CSF (n=50) of newly diagnosed paediatric patients with standard-risk ALL who received three intramuscular doses of Oncaspar (2,500 Units/m² BSA), one each during induction and two during delayed intensification treatment phases. A reduction in serum asparagine concentration was evident by the 4th day after the first Induction dose and reached an apparent nadir by the 10th day after the dose. Serum asparagine concentrations of approximately 1 μM persisted for approximately 3 weeks. Asparagine concentration fell to <3 μM when asparaginase activity was >0.1 U/mL. CSF asparagine of 2.3 μM pre-treatment fell to 1.1 μM on Day 7 and 0.6 μM on Day 28 of Induction (see Clinical efficacy and safety).
In Study AALL07P4, the PD effect of Oncaspar was assessed in 47 evaluable subjects with high risk B-precursor ALL who received IV doses of Oncaspar 2,500 U/m² BSA during the Induction and Consolidation phases. Plasma L-asparagine concentrations were depleted to below the assay limit of quantification within 24 hours following the Induction and first Consolidation dose of Oncaspar and depletion was sustained for approximately two weeks. CSF asparagine concentrations were reduced by the 4th day following the Induction dose, and remained largely undetectable by the 18th day after dosing.
Based on results from these two studies, a 2,500 U/m² BSA dose of Oncaspar administered IM (CCG-1962) and IV (AALL07P4) provides maintenance of L-asparagine depletion for approximately two weeks following dosing.
Clinical efficacy and safety
Oncaspar efficacy and safety were evaluated on the basis of three clinical studies using Oncaspar solution for injection/infusion in the first line treatment of ALL: Study CCG-1962 in standard risk ALL patients; Study AALL07P4 in high risk ALL patients; Study DFCI 11-001 enrolled both standard and high-risk ALL patients.
Oncaspar efficacy in ALL in patients with relapse/refractory disease and a history of prior clinical allergic reaction to native E. coli L-asparaginase was based on a pool of 94 patients from six open-label studies [ASP-001, ASP-201A, ASP-302, ASP-304, ASP-400 and ASP-001C/003C].
First-Line (ALL patients non-hypersensitive to native E. coli L-asparaginase)
The safety and efficacy of Oncaspar was evaluated in an open-label, multicentre, randomised, active-controlled study (StudyCCG-1962). In this study, 118 paediatric patients aged 1 to 9 years with previously untreated standard-risk ALL were randomised 1:1 to Oncaspar or native E. coli L-asparaginase as part of combination therapy. Oncaspar was administered intramuscularly at a dose of 2,500 Units/m² BSA on Day 3 of the 4-week Induction phase and on Day 3 of each of two 8-week Delayed Intensification (DI) phases. Native E. coli L-asparaginase was administered intramuscularly at a dose of 6,000 Units/m² BSA three times weekly for a total of 9 doses during induction and for a total of 6 doses during each delayed intensification phase.
The primary determination of efficacy was based on demonstration of similar asparagine depletion (magnitude and duration) in the Oncaspar and native E. coli L-asparaginase arms. The protocol-specified goal was achievement of asparagine depletion to a serum concentration of ≤1 μM. The proportion of patients with this level of depletion was similar between the 2 study arms during all 3 phases of treatment at the protocol-specified time points.
In all phases of treatment, serum asparagine concentrations decreased within 4 days of the first dose of asparaginase in the treatment phase and remained low for approximately 3 weeks for both Oncaspar and native E. coli L-asparaginase arms. Serum asparagine concentrations during the induction phase are shown in Figure 1. The patterns of serum asparagine depletion in the 2 delayed intensification phases are similar to the pattern of serum asparagine depletion in the induction phase.
Figure 1: Mean (± standard error) serum asparagine during Study CCG-1962 induction phase:
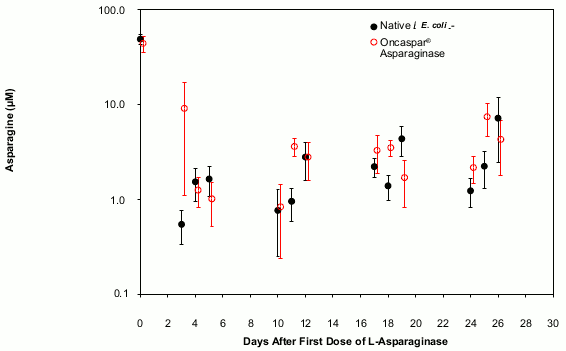
Note: Oncaspar (2,500 Units/m² BSA intramuscular) was administered on Day 3 of the 4-week induction phase. Native E. coli L-asparaginase (6,000 Units/m² BSA intramuscular) was administered 3 times weekly for 9 doses during induction.
CSF asparagine concentrations were determined in 50 patients during the induction phase. CSF asparagine decreased from a mean pre-treatment concentration of 3.1 μM to 1.7 μM on Day 4 ± 1 and 1.5 μM at 25 ± 1 days after administration of Oncaspar. These findings were similar to those observed in the native E. coli L-asparaginase treatment arm.
Event-Free Survival (EFS) for the Oncaspar and native E. coli L-asparaginase arms are summarised in Table 2, Study CCG-1962 was not designed to evaluate differences in EFS rates.
Table 2. Event-free survival rate at 3, 5 and 7 years (Study CCG-1962):
| Oncaspar | εγγενής L-ασπαραγινάση από E. coli | |
|---|---|---|
| 3-Year EFS Rate % (95% CI) | 83 (73, 93) | 79 (68, 90) |
| 5-Year EFS Rate, % (95% CI) | 78 (67, 88) | 73 (61, 85) |
| 7-Year EFS Rate, % (95% CI) | 75 (63, 87) | 66 (52, 80) |
In Study CCG-1962 , the most common adverse reactions were infections, including two life-threatening infections (1 patient in each arm). In general, incidence and type of adverse reactions Grade 3 and 4 were similar between the two treatment groups. Two patients in the Oncaspar arm had allergic reactions during Delayed Intensification (DI) DI #1 (Grade 1 allergic reaction and Grade 3 hives).
A pilot study was conducted for newly diagnosed patients from 1 to <31 years of age with high risk B-precursor ALL (Study AALL07P4). This was an open label, controlled, randomised study comparing an investigational pegylated asparaginase product to Oncaspar as a component of multi-agent chemotherapy in the first line treatment of ALL. White blood cell (WBC) criteria were: a) Age 1-10 years: WBC ≥50,000/μL; b) Age 10-30 years: Any WBC; c) Prior steroid therapy: Any WBC. Patients were not allowed prior cytotoxic chemotherapy with the exception of steroids and intrathecal cytarabine. A total of 166 patients were enrolled in this study; 54 patients were randomised to treatment with 2,500 U/m² BSA Oncaspar and 111 patients were randomised to the investigational pegylated asparaginase product. Oncaspar was administered intravenously at the dose of 2,500 Units/m² BSA during Induction, Consolidation, Delayed Intensification, and Interim Maintenance phases in patients with high-risk ALL receiving augmented Berlin-Frankfurt-Münster therapy. The percentage of patients in the Oncaspar treatment arm with evaluable minimal residual disease (MRD) negative status (<0.1% leukaemia cells in bone marrow) at Day 29 of Induction was 80% (40/50). At 4-years, the EFS and overall survival (OS) for the Oncaspar treatment arm were 81.8% [95% CI 62.9-91.7%] and 90.4% [95% CI 78.5-95.9%], respectively. Overall, in the group receiving Oncaspar, the rate of all grade hypersensitivity was 5.8%, anaphylactic reactions was 19.2%, and pancreatitis 7.7%. Grade 3 or higher febrile neutropenia was 15.4%.
Study DFCI 11-001, conducted by the Dana-Farber Cancer Institute (DFCI), is an ongoing, active-controlled, randomised multicentre study of an intravenous investigational pegylated asparaginase product versus Oncaspar, in children and adolescents aged 1 to <22 years with newly diagnosed ALL treated with a DFCI ALL consortium therapeutic backbone. A total of 239 patients were randomised, 237 of whom were treated with study drug (146 male and 91 female), of these, 119 patients (115 with a diagnosis of ALL) were treated with Oncaspar 2500 U/m². Treatment was administered during Induction (Day 7), and then every 2 weeks for a total of 30 weeks post-Induction therapy. Randomisation of patients was stratified based on risk group (standard/high/very high risk), including both B- and T-cell ALL. The percentage of patients in the Oncaspar arm with evaluable Low End-Induction MRD (<0.001 detectable disease) at Day 32 was 87.9% (80/91). The One-year EFS was 98.0 [95%CI 92.3, 99.5]; the One-year OS was 100 [95% CI 100, 100] in this study.
ALL patients hypersensitive to native E. coli L-asparaginase
Six open-label studies evaluated Oncaspar in relapse/refractory haematological diseases. In these studies a total of 94 patients with ALL diagnosis with a history of prior clinical allergic reaction to native E. coli L-asparaginase were exposed to Oncaspar. One patient received Oncaspar doses of 250 and 500 Units/m² BSA intravenously. The remaining patients were treated with 2,000 or 2,500 U/m² BSA administered intramuscularly or intravenously. Patients received Oncaspar as a single agent or in combination with multi-agent chemotherapy. Overall, from five studies analysed based on 65 ALL patients exposed to Oncaspar using the highest therapeutic response during the entire study, complete remission was observed in 30 patients (46%), partial remission in 7 patients (11%) and haematological improvement in 1 patient (2%). In the other study, with 29 hypersensitive ALL patients exposed to Oncaspar, 11 patients were evaluated for response during induction. Of these, 3 patients (27%) achieved complete remission, 1 patient (9%) had partial remission, 1 patient (9%) had haematologic improvement and 2 patients (18%) had therapeutic efficacy. Therapeutic efficacy was defined as a clinical improvement which did not meet the criteria for other beneficial outcomes. During the maintenance phase, 19 patients were evaluated, with 17 patients (89%) achieving complete remission, and 1 patient (5%) with therapeutic efficacy.
Pharmacokinetic properties
Oncaspar pharmacokinetic properties were based on asparaginase activity measured by an enzymatic assay after IM (CCG-1962) and IV (AALL07P4, DFCI 11-001) administration.
In Study CCG-1962, mean asparaginase activity reached peak value of 1 U/mL on Day 5 after the injection. The mean half-life after absorption from the injection site was 1.7 days and the elimination half-life was 5.5 days. The volume of distribution at steady-state and clearance were estimated at 1.86 L/m² and 0.169 L/m² per day, respectively.
In Study AALL07P4, PK parameters after a single 2,500 U/m² IV dose during Induction were calculated by noncompartmental PK analysis from sequential plasma samples and are depicted in Table 3 (see section 5.1). The Cmax and AUC of Oncaspar trended lower in males, subjects with larger BMI, and subjects >10 years. During Induction, following a single IV dose of Oncaspar 2,500 U/m², asparaginase activity ≥0.1 U/mL was sustained for up to 18 days post-dose in 95.3% of subjects.
Table 3. Pharmacokinetic Parameters After a Single IV Dose of Oncaspar 2,500 U/m² BSA During Induction (N=47; Study AALL07P4):
| PK Parameters | Arithmetic Mean (SD) |
|---|---|
| Cmax (mU/ml)* | 1638 (459.1) |
| Tmax (hr)* | 1,25 (1.08, 5.33)† |
| AUC0-t (mU·day/ml)* | 14810 (3555) |
| AUC0–∞ (mU·day/ml)ǂ | 16570 (4810) |
| t1/2 (day)ǂ | 5.33 (2.33) |
| CL (l/day)ǂ | 0,2152 (0,1214) |
| Vss (l)ǂ | 1.95 (1.13) |
In Study DFCI 11-001, assessments of asparaginase activity were performed following a single IV dose of Oncaspar 2,500 U/m² BSA during Induction, and every two weeks during post-Induction (see section 5.1). During Induction, plasma asparaginase activity ≥0.1 U/mL was sustained in 93.5% of subjects 18 days after administration. During the post-Induction phase, a nadir (trough) asparaginase activity above 0.4 U/mL was sustained in 100% of subjects from Week 7 up until Week 25. These results indicate that, when Oncaspar 2,500 U/m² BSA is administered as single and repeated doses every two weeks, clinically relevant asparaginase activity is sustained over the entire dosing interval (i.e., two weeks).
Patients with newly diagnosed ALL received a single IM injection of Oncaspar (2,500 U/m² BSA) or native asparaginase from E. coli (25,000 U/m² BSA) or from Erwinia (25,000 U/m² BSA). The plasma elimination half-life of Oncaspar was statistically significantly longer (5.7 days) than the plasma elimination half-lives of the native asparaginases from E. coli (1.3 days) and Erwinia (0.65 days). The immediate cell death of leukaemic cells in vivo, measured by rhodamine fluorescence, was the same for all three L-asparaginase preparations.
ALL patients with several relapses were treated either with Oncaspar or with native asparaginase from E. coli as part of an induction therapy. Oncaspar was given IM in a dose of 2,500 U/m² BSA on days 1 and 15 of induction. The mean plasma half-life of Oncaspar was 8 days in non-hypersensitive patients (AUC 10.35 U/ml/day), and 2.7 days in hypersensitive patients (AUC 3.52 U/ml/day).
Specific populations
The controlled studies were not designed to formally evaluate the pharmacokinetics of Oncaspar in specific populations. A population pharmacokinetic evaluation of Oncaspar based on data obtained from Studies AALL07P4 (IV), DFCI 11-001 (IV), and CCG-1962 (IM) identified that clearance (linear and saturable) increased approximately proportional to BSA and volume of distribution increased slightly more proportional to BSA. No statistically significant differences in PK characteristics between male and female subjects were identified in this analysis.
The impact of renal and hepatic impairment on the PK of Oncaspar has not been evaluated. As pegaspargase is a protein with a high molecular weight, it is not excreted renally, and no change of pharmacokinetic of Oncaspar in patients with renal impairment is foreseen.
Since the proteolytic enzymes responsible for Oncaspar metabolism are ubiquitously distributed in tissues the exact role of the liver is unknown: however any decrease in liver function is not expected to present clinical relevant problems in the use of Oncaspar.
There are no data available for elderly patients.
Preclinical safety data
Acute toxicity
Only very high doses of pegaspargase given to mice intraperitoneally as a single dose (25,000–100,000 U/kg body weight) caused the death of 14% of all treated mice. Mild hepatotoxicity was observed with the same dosages. Adverse reactions were loss of body weight, piloerection and reduced activity. Reduced splenic weight might be a sign of potential immunosuppressant effect of the treatment.
Pegaspargase was well tolerated both in rats and dogs when administered intravenously in single dose up to 500 U/kg body weight.
Repeated dose toxicity
A 4-week study in rats treated with a dose of pegaspargase of 400 U/kg/day intraperitoneal resulted in a fall in food intake and body weight compared to the control group.
A 3-month study in mice with pegaspargase at doses up to 500 U/kg intraperitoneal or intramuscular resulted in slight hepatocellular changes only at the highest intraperitoneal dose.
A temporary suppression in body weight gains and a temporary reduction in total leukocyte counts were observed in dogs which were treated with pegaspargase 1200 U/kg weekly for 2 weeks. Increased serum glutamic pyruvic transaminase activity also occurred in one out of four dogs.
Immunogenicity
No immunogenic response was detected in a 12-week study in mice in which pegaspargase was administered weekly at the dose of 10.5 U/mouse intramuscular or intraperitoneally.
Reproductive toxicity
No studies of reproductive toxicity were conducted with pegaspargase.
Embryotoxicity studies with L-asparaginase have given evidence of teratogenic potential in rats treated from day 6 to 15 of gestation with a No Observed Effect Level (NOEL) for teratogenic effects at 300 U/kg intravenous. In rabbits doses of 50 or 100 U/kg intravenous on days 8 and 9 of gestation induced viable fetuses with congenital malformations: no NOEL has been determined. Multiple malformations and embryolethal effects were observed with doses in the therapeutic range. Investigations of the effect on fertility and peri- and postnatal development were not conducted.
Carcinogenicity, mutagenicity, fertility
Long-term investigations of carcinogenicity or studies of the effect on fertility in animals were not conducted with pegaspargase.
Pegaspargase was not mutagenic in the Ames test using Salmonella typhimurium strains.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.