ORGOVYX Film-coated tablet Ref.[50889] Active ingredients: Relugolix
Source: European Medicines Agency (EU) Revision Year: 2023 Publisher: Accord Healthcare S.L.U., World Trade Center, Moll de Barcelona, s/n, Edifici Est 6ª planta, 08039 Barcelona, Spain
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Endocrine therapy, other hormone antagonists and related agents
ATC code: L02BX04
Mechanism of action
Relugolix is a nonpeptide GnRH receptor antagonist that competitively binds to GnRH receptors in the anterior pituitary gland preventing native GnRH from binding and signalling the secretion of luteinizing hormone (LH) and follicle-stimulating hormone (FSH). Consequently, the production of testosterone from the testes is reduced. In humans, FSH and LH concentrations rapidly decline upon initiating treatment with Orgovyx and testosterone concentrations are suppressed to below physiologic concentrations. Treatment is not associated with the initial increases in FSH and LH concentrations and subsequently testosterone ("potential symptomatic flare") observed upon initiation of treatment with a GnRH analogue. Following discontinuation of treatment, pituitary and gonadal hormone concentrations return to physiologic concentrations.
Clinical efficacy and safety
The safety and efficacy of Orgovyx was evaluated in HERO, a randomised, open-label study in adult men with androgen-sensitive advanced prostate cancer requiring at least 1 year of androgen deprivation therapy and who were not candidates for surgical or radiation therapy with curative intent. Eligible patients had either evidence of biochemical (PSA) or clinical relapse following local primary intervention with curative intent and were not candidates for salvage surgery, had newly diagnosed androgen-sensitive metastatic disease, or had advanced localized disease unlikely to be cured by primary intervention with either surgery or radiation. Eligible patients had to have an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1. Patients with disease progression during the treatment period were encouraged to remain on study and, if indicated, may have received radiotherapy as prescribed by the investigator. If PSA levels rose, patients were allowed to receive enzalutamide after the confirmation of PSA progression or docetaxel during the study.
The primary efficacy outcome measure was medical castration rate defined as achieving and maintaining serum testosterone suppression to castrate levels (<50 ng/dL) by day 29 through 48 weeks of treatment, plus non-inferiority of relugolix compared to leuprorelin was assessed (see Table 2). Other key secondary endpoints included castration rates on day 4 and 15, castration rates with testosterone <20 ng/dL at day 15, and PSA response rate at day 15 (see Table 3).
A total of 934 patients were randomised to receive Orgovyx or leuprorelin in a 2:1 ratio for 48 weeks:
a) Orgovyx at a loading dose of 360 mg on the first day followed by daily doses of 120 mg orally.
b) Leuprorelin 22.5 mg injection (or 11.25 mg in Japan, Taiwan, and China) subcutaneously every 3 months. Leuprorelin acetate 11.25 mg every 3 months is a dosage regimen that is not recommended for this indication in the European Union.
The population (N=930) across both treatment groups had a median age of 71 years (range 47 to 97 years). The ethnic/racial distribution was 68% White, 21% Asian, 4.9% Black, and 5% other. Disease stage was distributed as follows: 32% metastatic (M1), 31% locally advanced (T3/4 NX M0 or any T N1 M0), 28% localized (T1 or T2 N0 M0), and 10% not classifiable.
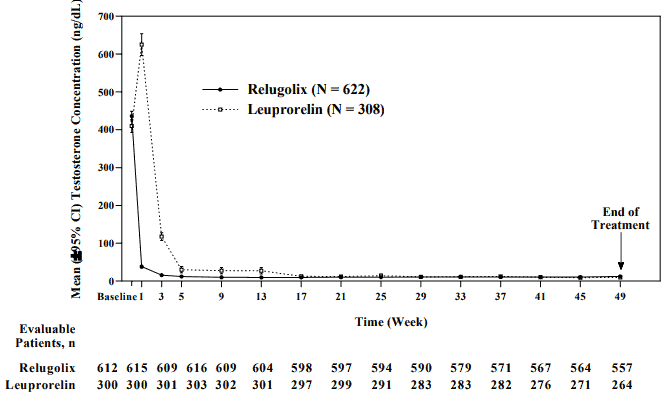
The primary efficacy results of Orgovyx to leuprorelin on achieving and maintaining serum testosterone at castrate levels (T<50 ng/dL) are shown in Table 2 and Figure 1. The baseline testosterone levels and the time-course of testosterone suppression by Orgovyx and leuprorelin during the 48 week treatment period are shown in Figure 2.
Table 2. Medical castration rates (testosterone concentrations <50 ng/dL) from week 5 day 1 (day 29) through week 49 day 1 (day 337) in HERO:
| Orgovyx 360/120 mg | Leuprorelin 22.5 or 11.5 mga | |
|---|---|---|
| No. treated | 622b | 308b |
| Responder rate (95% CI)c | 96.7% (94.9%, 97.9%) | 88.8% (84.6%, 91.8%) |
| Difference from leuprorelin (95% CI) | 7.9% (4.1%, 11.8%)d p-value <0.0001 |
a 22.5 mg dosed in Europe and North America; 11.25 mg dosed in Asia. The castration rate of the subgroup of patients receiving 22.5 mg leuprorelin (n=264) was 88.0% (95% CI: 83.4%, 91.4%).
b Two patients in each arm did not receive the study treatment and were not included.
c Kaplan-Meier estimates within group.
d Non-inferiority was tested with a margin of -10%.
Figure 1. Cumulative incidence of testosterone concentrations <50 ng/dL in HERO:
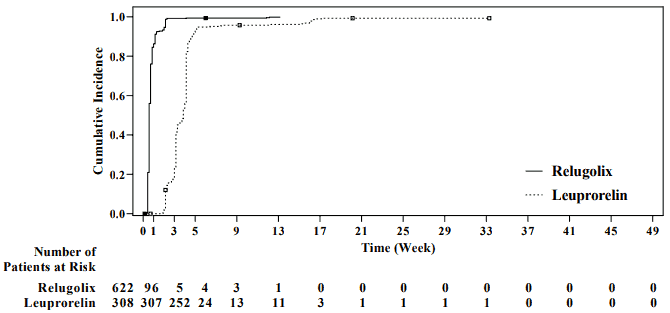
Figure 2. Testosterone concentrations from baseline to week 49 (mean and 95% CI) in HERO:
A summary of the results of the key secondary endpoints are shown in Table 3.
Table 3. Summary of key secondary endpoints:
| Secondary endpoint | Orgovyx (N=622) | Leuprorelin (N=308) | p-Value |
|---|---|---|---|
| Cumulative probability of testosterone suppression to <50 ng/dL prior to dosing on day 4 | 56.0 | 0.0 | <0.0001 |
| Cumulative probability of testosterone suppression to <50 ng/dL prior to dosing on day 15 | 98.7 | 12.1 | <0.0001 |
Proportion of patients with PSA response at Day 15
followed with confirmation at day 29
79.4 19.8 <0.0001
Cumulative probability of testosterone suppression to
< 20 ng/dL prior to dosing on day 15
78.4 1.0 <0.0001
Abbreviations: PSA = prostate-specific antigen.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with Orgovyx in all subsets of the paediatric population in treatment of advanced hormone-sensitive prostate cancer (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
After oral administration of a single 360 mg loading dose, the mean (± standard deviation [± SD]) of AUC0-24 and Cmax of relugolix were 985 (± 742) ng.hr/mL and 215 (± 184) ng/mL, respectively. After administration of a 120 mg dose once daily, the mean (± SD), Cmax, Cavg (average plasma concentration over the 24-hour dosing interval), and Ctrough of relugolix at steady-state were 70 (± 65) ng/mL, 17.0 (± 7) ng/mL and 10.7 (± 4) ng/mL, respectively.
The accumulation of exposure to relugolix upon once daily administration of a 120-mg dose of relugolix is approximately 2-fold. After once daily administration of relugolix following a 360-mg loading dose on the first day of administration, steady state of relugolix is achieved by day 7.
Absorption
The absorption of relugolix after oral administration is primarily mediated by intestinal P-gp, for which relugolix is a substrate. After oral administration, relugolix is rapidly absorbed, reaching quantifiable concentration by 0.5 hours post-dose followed by one or more subsequent absorption peaks. The median (range) time to Cmax (tmax) of relugolix is 2.25 hours (0.5 to 5.0 hours). The absolute bioavailability of relugolix is 11.6%.
After administration of a single 120-mg dose of relugolix following consumption of a high-calorie, high-fat meal (approximately 800 to 1 000 calories with 500, 220, and 124 from fat, carbohydrate, and protein, respectively), the AUC0-∞ and Cmax were decreased 19% and 21%, respectively. The decreases in exposure to relugolix with food are not considered to be clinically meaningful and therefore Orgovyx may be administered without regard to food (see section 4.2).
Distribution
Relugolix is 68 to 71% bound to plasma proteins, primarily to albumin and to a lesser extent to α1-acid glycoprotein. The mean blood-to-plasma ratio is 0.78. Based on the apparent volume of distribution (Vz), relugolix distributes widely to tissues. The estimated volume of distribution at steady state (Vss) is 3 900 L.
Biotransformation
In vitro studies indicate that the primary CYP enzymes contributing to the overall hepatic oxidative metabolism of relugolix were CYP3A4/5 (45%) > CYP2C8 (37%) > CYP2C19 (<1%) with the oxidative metabolites, Metabolite-A and Metabolite-B, formed by CYP3A4/5 and CYP2C8, respectively.
Elimination
Once absorbed, approximately 19% of relugolix is eliminated as unchanged active substance in the urine and approximately 80% is eliminated through multiple biotransformation pathways, including CYP3A and CYP2C8 and multiple other minor metabolic pathways, with a minor contribution from biliary secretion of unchanged medicinal product and/or metabolites. Approximately 38% of the administered dose is excreted as metabolites (other than Metabolite-C) in the faeces and urine. Metabolite-C, which is formed by intestinal microflora, is the primary metabolite in faeces (51%) and further reflects non-absorbed drug.
Linearity/non-linearity
Relugolix is associated with greater than dose-proportional increases in exposure at doses below approximately 80 mg, which is consistent with the dose-dependent saturation of intestinal P-gp and the corresponding decreasing contribution of intestinal P-gp efflux to the oral bioavailability of relugolix as the dose is increased. Upon saturation of intestinal P-gp, a greater proportion of the absorption of relugolix is governed by passive diffusion, and the exposure to relugolix increases in proportion to dose within the 80- to 360-mg dose range. The saturation of intestinal P-gp with higher doses of relugolix is demonstrated by the dose-related increases in exposure to relugolix associated with erythromycin, a strong P-gp inhibitor (and moderate CYP3A inhibitor), where the increases in exposure was less for a 120-mg dose compared with lower doses of relugolix (20 or 40 mg) (see section 4.5).
Special populations
Population PK (PopPK) and PopPK/PD analyses suggest that there are no clinically meaningful differences in exposure of relugolix or testosterone concentrations based on age, race or ethnicity, body size (body weight or body mass index) or stage of cancer.
Renal impairment
Based upon the dedicated renal impairment studies with 40 mg relugolix, the exposure to relugolix (AUC0-t) was increased by 1.5-fold in patients with moderate renal impairment and by up to 2.0-fold in patients with severe renal impairment as compared to subjects with normal renal function. The increases in patients with moderate renal impairment are not considered to be clinically meaningful. With respect to patients with severe renal impairment, caution is warranted upon once daily administration of a 120-mg dose of relugolix (see section 4.4).
The effect of end stage renal disease with or without haemodialysis on the pharmacokinetics of relugolix has not been evaluated. The amount of relugolix removed by haemodialysis is unknown.
Hepatic impairment
After administration of a single 40-mg dose of relugolix to patients with mild or moderate hepatic impairment, the total exposure to relugolix (AUC0-∞) was decreased by 31% or was comparable, respectively, compared to subjects with normal hepatic function. The mean elimination half-life of relugolix in patients with mild or moderate hepatic impairment and healthy control subjects was comparable.
No dose adjustment for Orgovyx in patients with mild or moderate hepatic impairment is required (see section 4.2). The effects of severe hepatic impairment on the pharmacokinetics of relugolix have not been evaluated.
5.3. Preclinical safety data
Non-clinical data based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, or carcinogenic potential reveal no special hazard for humans beyond those discussed below.
In human GnRH-receptor knock-in male mice, oral administration of relugolix decreased prostate and seminal vesicle weights at doses ≥3 mg/kg twice daily for 28 days. The effects of relugolix were reversible, except for testis weight, which did not fully recover within 28 days after drug withdrawal. These effects in knock-in male mice are likely associated with the pharmacodynamics of relugolix; however, the relevance of these findings to humans is unknown. In a 39-week repeat dose toxicity study in monkeys, there were no significant effects on male reproductive organs at oral relugolix doses up to 50 mg/kg/day (approximately 36 times the human exposure at the recommended dose of 120 mg daily based on AUC). Relugolix (doses of ≥1 mg/kg) suppressed LH concentrations in castrated male cynomolgus monkeys; however, the suppressive effect of relugolix on LH and sex hormones was not evaluated in the 39-week toxicity study in intact monkeys. Therefore, the relevance of the lack of effect on reproductive organs in intact male monkeys to humans is unknown.
In pregnant rabbits orally dosed with relugolix during the period of organogenesis, spontaneous abortion and total litter loss were observed at exposure levels (AUC) less than that achieved at the recommended human dose of 120 mg/day. No effects on embryofoetal development were observed in rats; however, relugolix does not interact significantly with GnRH receptors in that species.
In lactating rats administered a single oral dose of 30 mg/kg radiolabelled relugolix on post-partum day 14, relugolix and/or its metabolites were present in milk at concentrations up to 10-fold higher than in plasma at 2 hours post-dose decreasing to low levels by 48 hours post-dose. The majority of relugolix-derived radioactivity in milk consisted of unchanged relugolix.
Environmental risk assessment studies have shown that relugolix may pose a risk for the aquatic compartment (see section 6.6).
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.