ORSERDU Film-coated tablet Ref.[51613] Active ingredients: Elacestrant
Source: European Medicines Agency (EU) Revision Year: 2023 Publisher: Stemline Therapeutics B.V., Basisweg 10, 1043 AP Amsterdam, The Netherlands
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Endocrine therapy, anti estrogen
ATC code: L02BA04
Mechanism of action
Elacestrant, a tetrahydronaphthalene compound, is a potent, selective and orally active estrogen receptor-α (ERα) antagonist and degrader.
Pharmacodynamic effects
Elacestrant inhibits the estradiol-dependent and independent growth of ERα-positive breast cancer cells, including models harbouring estrogen receptor 1 (ESR1) gene mutations. Elacestrant displayed potent antitumor activity in patient derived xenograft models previously exposed to multiple endocrine therapies, harbouring wild type ESR1 or ESR1 gene mutations in the ligand binding domain.
In patients with ER+ advanced breast cancer with a median of 2.5 prior lines of endocrine therapy, dosed with elacestrant dihydrochloride 400 mg (345 mg of elacestrant) daily, median reduction in tumour 16α-18F-fluoro-17β-estradiol (FES) uptake from baseline to Day 14 was 88.7% demonstrating reduced ER availability and antitumor activity measured by FES-PET/CT in patients with prior endocrine therapies.
Clinical efficacy and safety
The efficacy and safety of ORSERDU in patients with ER+/HER2- advanced breast cancer following prior endocrine therapy in combination with a CDK4/6 inhibitor was evaluated in RAD1901-308, a randomised, open-label, active-controlled, multicenter trial which compared ORSERDU with standard of care (SOC) (fulvestrant for patients who received prior aromatase inhibitors in the metastatic setting or aromatase inhibitors for patients who received fulvestrant in the metastatic setting). Eligible patients included post-menopausal women and men whose disease had relapsed or progressed on at least 1 and no more than 2 prior lines of endocrine therapy. All patients were required to have Eastern Cooperative Oncology Group (ECOG)performance status of 0 or 1, and evaluable lesions per Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1, i.e., measurable disease or bone only disease with evaluable lesions. Prior endocrine therapy must have included a combination with CDK4/6 inhibitor therapy and no more than 1 prior line of cytotoxic chemotherapy for metastatic breast cancer. Patients were required to be appropriate candidates for endocrine monotherapy. Patients with presence of symptomatic metastatic visceral disease, patients with cardiac comorbidity, and patients with severe hepatic impairment were excluded.
A total of 478 patients were randomised 1:1 to daily oral administration of 400 mg of elacestrant dihydrochloride (345 mg of elacestrant) or standard of care (SOC) (239 on elacestrant and 239 on SOC), including a total of 228 patients (47.7%) with ESR1 mutations at baseline (115 patients on elacestrant and 113 patients on SOC). Among the 239 patients randomised to the SOC arm, 166 received fulvestrant, and 73 received an aromatase inhibitor that included anastrozole, letrozole or exemestane. Randomisation was stratified by ESR1 mutations status (ESR1-mut vs ESR1-mut-nd [no ESR1 mutations detected]), prior treatment with fulvestrant (yes vs no), and visceral metastasis (yes vs no). ESR1 mutational status was determined by blood circulating tumor deoxyribonucleic acid (ctDNA) using the Guardant360 CDx assay and was limited to ESR1 missense mutations in the ligand binding domain (between codons 310 to 547).
The median age of patients (ORSERDU vs standard of care) at baseline was 63.0 years (range of 24-89) vs 63.0 (range of 32-83) and 45.0% were over 65 (43.5 vs 46.4). Most patients were women (97.5% vs 99.6%) and most patients were white (88.4% vs 87.2%), followed by Asian (8.4% vs 8.2%), Black or African American (2.6% vs 4.1%), and Other/Unknown (0.5% vs 0.5%). Baseline ECOG performance status was 0 (59.8% vs 56.5%), 1 (40.2% vs 43.1%) or >1 (0% vs 0.4%). Patient demographics for those with ESR1-mutated tumors were generally representative of the broader study population. The median duration of exposure to ORSERDU was 2.8 months (range: 0.4 to 24.8).
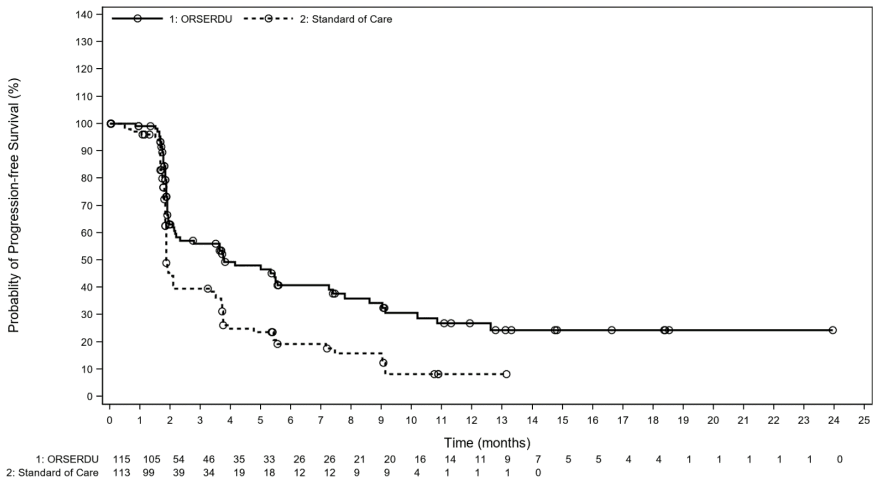
The primary efficacy endpoint was progression-free survival (PFS) as assessed by IRC (Independent Review Committee) in all patients, i.e., including patients with an ESR1 mutation, and in patients with ESR1 mutations. A statistically significant PFS benefit was observed in all patients with a median PFS of 2.79 months in the Orserdu arm as compared with 1.91 months in the standard of care arm (HR= 0.70, 95% CI: 0.55, 0.88). Efficacy results are presented in Table 4 and Figure 1 for patients with ESR1 mutations.
Table 4. Efficacy results among patients with ESR1 mutations (evaluated by a blinded imaging review committee):
| ORSERDU | Standard of care | |
|---|---|---|
| Progression-free survival (PFS) | N=115 | N=113 |
| Number of PFS events, n (%) | 62 (53.9) | 78 (69.0) |
| Median PFS months* (95% CI) | 3.78 (2.17, 7.26) | 1.87 (1.87, 2.14) |
| Hazard ratio** (95% CI) | 0.546 (0.387, 0.768) | |
| p-value (stratified log-rank) | 0.0005 | |
| Overall survival (OS) | N=115 | N=113 |
| Number of OS events, n (%) | 61 (53) | 60 (53.1) |
| Median OS months* (95% CI) | 24.18 (20.53, 28.71) | 23.49 (15.64, 29.90) |
| Hazard ratio** (95% CI) | 0.903 (0.629, 1.298) | |
CI=confidence interval; ESR1=estrogen receptor 1; PFS=progression-free survival.
* Kaplan-Meir estimate; 95% CI based on the Brookmeyer-Crowley method using a linear transformation.
** From a Cox proportional hazards model stratified by prior treatment with fulvestrant (yes vs no), and visceral metastasis (yes vs no).
Data cut-off dates are 06 September 2021 for PFS and 02 September 2022 for OS.
Figure 1. PFS in patients with an ESR1 mutation (evaluated by a blinded imaging review committee):
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with ORSERDU in all subsets of the paediatric population in breast cancer (see section 4.2).
5.2. Pharmacokinetic properties
The elacestrant oral bioavailability is approximately 10%. Steady state is reached by Day 6 following once daily dosing. Cmax and AUC increase slightly more than proportional to dose for doses ≥50 mg (salt form).
Absorption
Following oral administration, elacestrant was rapidly absorbed, reaching Cmax within 1-4 hours. The geometric mean Cmax was 52.86 ng/mL (35.2% coefficient of variation [CV%]) and AUCinf was 1566 ng*h/mL (38.4% CV) after single dose administration of 345 mg of elacestrant in fed conditions. At steady state, the median [min, max] plasma concentration at 4h post-dose (C4h) and AUC are predicted to be 108 ng/mL [27.5–351] and 2190 ng*h/mL [461–8470], respectively.
Effect of food
Administration of elacestrant 345 mg tablet with a high-fat high-calorie meal increased Cmax and AUC by 40% and 20%, respectively, as compared to fasted administration. When the tablet was coadministered with a light meal, Cmax and AUC increased in a similar fashion, i.e., by 30 and 20%, respectively. Ingestion with food may reduce gastrointestinal adverse effects.
Distribution
Plasma protein binding of elacestrant is >99% and independent of concentration and hepatic impairment status. Elacestrant penetrates the blood brain barrier in a dose-dependent manner. Following once daily administration of elacestrant for 7 consecutive days, median concentrations of elacestrant in the cerebrospinal fluid were 0.0966 ng/mL and 0.155 ng/mL at the doses of 200 and 500 mg, respectively.
Based on population pharmacokinetic analysis, elacestrant is extensively distributed in the tissues with an apparent peripheral volume of distribution of 5411 L. The apparent central volume of distribution of elacestrant at steady state is 422 L.
Biotransformation
Elacestrant was a minor (<10% of plasma radioactivity) component in human plasma. 4-[2-(Ethylamino)ethyl]benzoic acid (EAEBA) glucuronide was a major human plasma metabolite (about 41% of plasma radioactivity). Elacestrant is primarily metabolised by CYP3A4 with a potential small contribution by CYP2A6 and CYP2C9.
Elimination
The half-life of elacestrant is predicted to be approximately 30 hours. After a single dose, the mean (% CV) clearance of elacestrant was 220.3 L/hr (38.4%). At steady state, the mean (% CV) clearance of elacestrant is predicted to be 186 L/hr (43.5%).
Following a single oral dose of 345 mg radiolabeled elacestrant, 81.5% (majority as unchanged) was recovered in feces and 7.53% (trace as unchanged) was recovered in urine. Elacestrant renal clearance is very low (≤2.3 mL/min) and it was eliminated by oxidative metabolism and fecal excretion.
Special populations
Effect of age, weight and gender
From analyses of population pharmacokinetic data in cancer patients, no dose adjustment is warranted based on body weight, age, and gender.
Hepatic impairment
The Cmax and AUC values were similar between subjects in the mild hepatic impairment group (ChildPugh A) and the normal hepatic function group upon single dose administration of elacestrant 176 mg. There were significant increases in AUC0–t (76%) and AUC0–∞ (83%) in the moderate hepatic impairment group (Child-Pugh B) compared to the normal hepatic function group. The Cmax values were similar between the normal and moderate impairment groups.
The geometric mean elimination half-life (t1/2) tended to increase with increasing severity of hepatic impairment. Elacestrant has not been studied in subjects with severe hepatic impairment (Child-Pugh C).
In PBPK modeling simulation of elacestrant at 345 mg, the steady state AUC and Cmax were predicted to increase by 2.14- and 1.92-fold, respectively, in subjects with moderate hepatic impairment compared to patients with normal hepatic function.
5.3. Preclinical safety data
Elacestrant displayed low acute toxicity. In repeated dose toxicity studies in rats and monkeys, the antiestrogenic activity of elacestrant was responsible for the effects seen, particularly in the female reproductive system, but also in other organs sensitive to hormones such as mammary gland, pituitary and testes. Sporadic emesis and diarrhoea were recorded in monkeys. In addition, in long-term studies (26 weeks in rats and 39 weeks in cynomolgus monkeys), increased vacuolation of the mucosal epithelium of the non-glandular stomach were observed in rats and vacuolated macrophage infiltrates in the small intestine were recorded in both rats and monkeys. In monkeys this effect occurred at a level of systemic exposure of about 70% of the human exposure.
Elacestrant showed no genotoxic potential in the Ames test, chromosomal aberrations in human lymphocytes and in the micronucleus assay in rats.
Fertility studies in animals have not been conducted. In repeated-dose toxicity studies effects related to fertility were observed in rat and monkey female reproductive tract; these effects occurred below human exposures at MRHD (maximum recommended dose). Decreased cellularity of Leydig cells in rat testes was also observed at exposure levels 2.7-fold higher than in humans.
In embryo-foetal development studies in rats, oral administration of elacestrant resulted in maternal toxicity (body weight loss, low food consumption, red vulvar discharge) and increased resorptions, increased post-implantation loss, and reduced number of live foetuses and foetal variations and malformations below human exposures at MHRD.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.