OXLUMO Solution for injection Ref.[10953] Active ingredients: Lumasiran
Source: European Medicines Agency (EU) Revision Year: 2020 Publisher: Alnylam Netherlands B.V., Antonio Vivaldistraat 150, 1083 HP Amsterdam, Netherlands
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: not yet assigned
ATC code: not yet assigned
Mechanism of action
Lumasiran is a double-stranded small interfering ribonucleic acid (siRNA) that reduces levels of glycolate oxidase (GO) enzyme by targeting the hydroxyacid oxidase 1 (HAO1) gene messenger ribonucleic acid (mRNA) in hepatocytes through RNA interference. Decreased GO enzyme levels reduce the amount of available glyoxylate, a substrate for oxalate production. This results in reduction of urinary and plasma oxalate levels, the underlying cause of disease manifestations in patients with PH1. As the GO enzyme is upstream of the deficient alanine: glyoxylate aminotransferase (AGT) enzyme that causes PH1, the mechanism of action of lumasiran is independent of the underlying AGXT gene mutation.
Clinical efficacy
The efficacy of lumasiran was studied in a randomised, double-blind, placebo-controlled clinical study in patients 6 years and older with PH1 (ILLUMINATE-A) and in a single-arm clinical study in patients less than 6 years of age with PH1 (ILLUMINATE-B).
ILLUMINATE-A
A total of 39 patients with PH1 were randomised 2:1 to receive subcutaneous doses of lumasiran or placebo during the 6-month double-blind, placebo-controlled period. Patients 6 years and older with an estimated glomerular filtration rate (eGFR) ≥30 mL/min/1.73 m² were enrolled, and received 3 loading doses of 3 mg/kg lumasiran or placebo administered once monthly, followed by quarterly maintenance doses of 3 mg/kg lumasiran or placebo (see section 4.2). After the 6-month double-blind treatment period, patients, including those originally assigned to placebo, entered an extension period with administration of lumasiran.
During the 6-month double-blind, placebo-controlled period, 26 patients received lumasiran, and 13 received placebo. The median age of patients at first dose was 14.9 years (range 6.1 to 61.0 years), 66.7% were male, and 76.9% were white. The median 24-hour urinary oxalate excretion corrected for body surface area (BSA) at baseline was 1.72 mmol/24 h/1.73 m² , the median spot urinary oxalate: creatinine ratio at baseline was 0.21 mmol/mmol, and the median plasma oxalate level at baseline was 13.1 µmol/L. Overall, 33.3% of patients had normal renal function (eGFR ≥90 mL/min/1.73 m²), 48.7% had mild renal impairment (eGFR of 60 to <90 mL/min/1.73 m²), and 18% had moderate renal impairment (eGFR of 30 to <60 mL/min/1.73 m²). Of the patients enrolled in the study, 84.6% reported a history of symptomatic renal stone events and 53.8% reported a history of nephrocalcinosis at baseline. The treatment arms were balanced at baseline with respect to age, urinary oxalate level, and eGFR.
The primary endpoint was the percent reduction from baseline in 24-hour urinary oxalate excretion corrected for BSA averaged over months 3 through 6. Lumasiran was associated with a statistically significant reduction of 65.4% in 24-hour urinary oxalate corrected for BSA, as compared to 11.8% in the placebo group, representing a difference of 53.5% (95% CI: 44.8, 62.3; p<0.0001). Consistent with the primary endpoint, a reduction of 60.5% was observed at month 6 in spot urinary oxalate: creatinine ratio in the lumasiran arm compared to an 8.5% increase in the placebo arm. Furthermore, patients treated with lumasiran had a rapid and sustained decrease in 24-hour urinary oxalate corrected for BSA, as shown in Figure 1.
Figure 1. ILLUMINATE-A: Percent change from baseline in 24-hour urinary oxalate corrected for BSA by month:
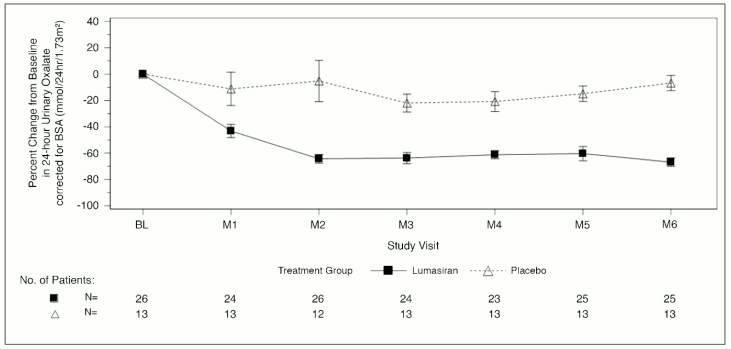
Abbreviations: BL = baseline; BSA = body surface area; M = month; SEM = standard error of mean. Results are plotted as mean (±SEM) of percent change from baseline.
At month 6, a higher proportion of lumasiran-treated patients achieved normal or near-normal levels of 24-hour urinary oxalate corrected for BSA (≤1.5×ULN) compared to placebo-treated patients, as shown in Table 3.
Table 3. ILLUMINATE-A: Secondary endpoint results over the 6-month double-blind, placebo-controlled period:
| Endpoints | Lumasiran (N=26) | Placebo (N=13) | Treatment difference (95% CI) | p-value |
|---|---|---|---|---|
| Proportion of patients with 24-hour urinary oxalate levels at or below ULN‡ | 0.5 (0.3, 0.7)§ | 0 (0, 0.2)§ | 0.5 (0.2, 0.7)¶ | 0.001# |
| Proportion of patients with 24-hour urinary oxalate levels at or below 1.5×ULN‡ | 0.8 (0.6, 1.0)§ | 0 (0, 0.2)§ | 0.8 (0.5, 0.9)¶ | <0.0001# |
| Percent reduction in plasma oxalate from baseline*Þ | 39.8 (2.9)† | 0.3 (4.3)† | 39.5 (28.9, 50.1) | <0.0001 |
Abbreviations: ULN = upper limit of normal; SEM = Standard Error of Mean
Results are based on liquid chromatography tandem mass spectrometry (LC-MS/MS) assay.
* The estimate based on the average of the least square mean of percent reduction at Months 3, 4, 5, and 6 using a mixed model for repeated measures.
† LS Mean (SEM).
‡ ULN=0.514 mmol/24 hr/1.73 m² for 24-hour urinary oxalate corrected for BSA.
§ 95% CI based on Clopper Pearson Exact confidence interval.
¶ Calculated using the Newcombe Method based on the Wilson Score.
# p-value is based on Cochran–Mantel–Haenszel test stratified by baseline 24-hour urinary oxalate corrected for BSA (≤1.70 vs >1.70 mmol/24 hr/1.73 m²).
Þ Analysed in 23 lumasiran and 10 placebo patients who had baseline levels that allowed for reduction to occur.
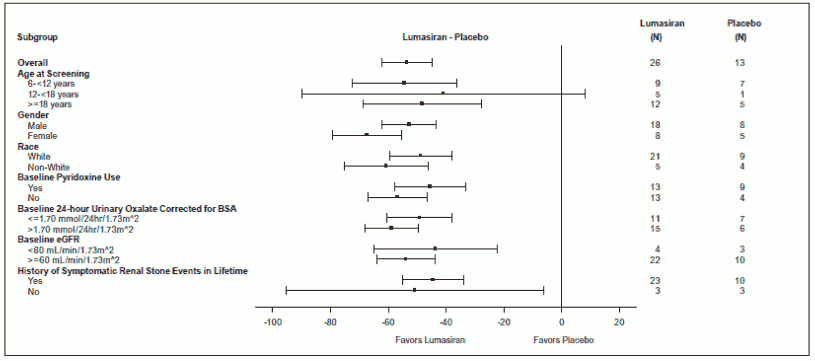
Reduction in 24-hour urinary oxalate corrected for BSA from baseline in patients with PH1 receiving lumasiran compared to placebo was similar across all pre-specified subgroups, including age, sex, race, renal impairment, baseline pyridoxine (vitamin B6) use, and history of symptomatic renal stone events (Figure 2).
Figure 2. ILLUMINATE-A: Percent change from baseline in 24-hour urinary oxalate corrected for BSA, subgroup analysis:
Reduced oxalate levels observed in the double-blind period were maintained through 12 months during the extension period of study.
eGFR and renal stone events (reported by events per 100 person-days) were assessed through the double-blind and extension periods for a total of 12 months. eGFR remained stable in patients administered lumasiran. In the lumasiran arm, the rate of renal stone events reported 12 months prior to consent was 0.87 (95% CI: 0.70, 1.08). The observed events during the double-blind period and the first 6 months of the extension period were 0.30 (95% CI: 0.17, 0.51) and 0.23 (95% CI: 0.13, 0.43), respectively. In the placebo arm, the rate of renal stone events reported 12 months prior to consent was 0.15 (95% CI: 0.07, 0.31) and the observed events during the double-blind period were 0.18 (95% CI: 0.07, 0.48). During the first 6 months of lumasiran treatment in the extension period a rate of 0.05 (95% CI: 0.01, 0.32) events were observed in patients previously receiving placebo. For nephrocalcinosis, data through the 6-month double-blind period are available. Of the 34 patients with baseline and month 6 renal ultrasounds, 3 of 22 in the lumasiran group showed improvement in nephrocalcinosis, and 1 of 12 in the placebo group showed worsening in nephrocalcinosis. None of the other lumasiran (n=19) or placebo-treated (n=11) patients exhibited a change in nephrocalcinosis.
ILLUMINATE-B
A total of 18 patients were enrolled and treated with lumasiran in an ongoing multi-center, single-arm study in patients with PH1 (ILLUMINATE-B). The study enrolled patients less than 6 years of age with an eGFR >45 mL/min/1.73 m² in patients 12 months of age and older, and normal serum creatinine in patients less than 12 months of age. In the 6-month primary analysis, at first dose, 3 patients were less than 10 kg, 12 were 10 kg to less than 20 kg, and 3 were 20 kg and above. The median age of patients at first dose was 51.4 months (range 4.0 to 74.0 months), 55.6% were female, and 88.9% were white. The median spot urinary oxalate: creatinine ratio at baseline was 0.47 mmol/mmol.
At month 6, patients treated with lumasiran achieved a reduction of 72.0% (95% CI: 66.4, 77.5) in spot urinary oxalate: creatinine ratio from baseline (averaged over months 3 through month 6), the primary endpoint. Lumasiran was associated with rapid, and sustained reductions in spot urinary oxalate: creatinine ratio (Figure 3), which were similar across all weight strata. The percent reduction in urinary oxalate excretion was consistent with data from ILLUMINATE-A.
Figure 3. ILLUMINATE-B: Percent change in spot urinary oxalate: creatinine ratio from baseline by month:
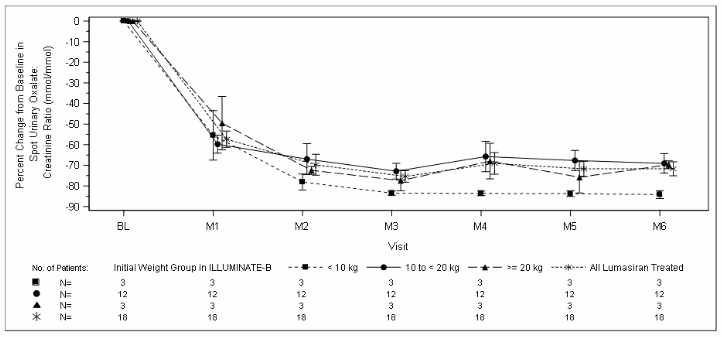
Nine patients achieved near normalisation (≤1.5×ULN), including 1 patient who achieved normalisation (≤ULN), at month 6 in spot urinary oxalate: creatinine ratio.
Furthermore, from baseline to month 6 (average of month 3 to month 6), a mean plasma oxalate reduction of 31.7% (95% CI: 23.9, 39.5) was observed. During the 6-month period, the eGFR remained stable, and 2 post-baseline renal stone events in 2 patients were reported, compared to 4 renal stone events in 3 patients in the 12-month period prior to consent. Fourteen of 18 patients had baseline nephrocalcinosis. Renal ultrasound data at month 6 indicated improvement in 8 patients, including 3 with bilateral improvement. None of the 18 patients had new onset or worsening nephrocalcinosis.
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with Oxlumo in one or more subsets of the paediatric population in hyperoxaluria (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Absorption
Following subcutaneous administration, lumasiran is rapidly absorbed with a median (range) time to reach maximum plasma concentration (tmax) of 4.0 (0.5 to 12.0) hours. In children and adults with PH1 ≥20 kg, the peak plasma concentration of lumasiran (Cmax) and area under the concentration curve from time zero to the last measurable concentration after dosing (AUC0-last) following the recommended lumasiran dose of 3 mg/kg were 529 (205 to 1130) ng/mL and 7400 (2890 to 10700) ng·h/mL, respectively. In children less than 20 kg, Cmax and AUC0-last of lumasiran following the recommended lumasiran dose of 6 mg/kg were 912 (523 to 1760) and 7960 (5920 to 13300). Lumasiran concentrations were measurable, up to 24 to 48 hours post-dose.
Distribution
In healthy adult plasma samples, the protein binding of lumasiran is moderate to high (77 to 85%) at clinically relevant concentrations. For an adult patient with PH1, the population estimate for the apparent central volume of distribution (Vd/F) for lumasiran is 4.9 L. Lumasiran primarily distributes to the liver after subcutaneous dosing.
Biotransformation
Lumasiran is metabolised by endo- and exonucleases to oligonucleotides of shorter lengths. In vitro studies indicate that lumasiran does not undergo metabolism by CYP450 enzymes.
Elimination
Lumasiran is primarily eliminated from plasma by hepatic uptake, with only 7 to 26% of the administered dose recovered in urine as lumasiran in the pooled data from healthy adult subjects and patients with PH1 >6 years of age. The mean (%CV) terminal plasma half-life of lumasiran is 5.2 (47.0%) hours. The population estimate for apparent plasma clearance was 26.5 L/h for a typical 70-kg adult. The mean renal clearance of lumasiran was minor and ranged from 2.0 to 3.4 L/h in paediatric and adult patients with PH1.
Linearity/non-linearity
Lumasiran exhibited linear to slightly nonlinear, time-independent pharmacokinetics in plasma following single subcutaneous doses ranging from 0.3 to 6 mg/kg and multiple doses of 1 and 3 mg/kg once monthly or 3 mg/kg quarterly. There was no accumulation of lumasiran in plasma after repeated once monthly or quarterly dosing.
Pharmacokinetic/pharmacodynamic relationship(s)
Plasma concentrations of lumasiran do not reflect the extent or duration of the pharmacodynamic activity of lumasiran. Rapid and targeted uptake of lumasiran by the liver results in rapid decline in plasma concentrations. In the liver, lumasiran exhibits a long half-life leading to maintenance of pharmacodynamic effect over the monthly or quarterly dosing interval.
Interactions
In vitro studies indicate that lumasiran is not a substrate or an inhibitor of cytochrome P450 (CYP) enzymes. Lumasiran is not expected to inhibit or induce CYP enzymes or modulate the activities of drug transporters.
Special populations
Elderly
No studies have been conducted in patients ≥65 years of age. Age was not a significant covariate in the pharmacokinetics of lumasiran.
Gender and race
In clinical studies, there was no difference in the plasma exposure or pharmacodynamics of lumasiran based on gender or race.
Hepatic impairment
No studies have been conducted in patients with hepatic impairment (see section 4.2). Limited pharmacokinetic data in patients with mild and transient elevations in total bilirubin (total bilirubin >1.0 to 1.5×ULN) showed comparable plasma exposure of lumasiran and similar pharmacodynamics as patients with normal hepatic function. Published literature show lower expression of the asialoglycoprotein receptors in the liver, i.e. the receptors responsible for lumasiran uptake, in patients with hepatic impairment. Nonclinical data suggest that this may not influence liver uptake or pharmacodynamics at therapeutic doses. The clinical relevance of these data is unknown.
Renal impairment
Patients with mild renal impairment (eGFR 60 to <90 mL/min/1.73 m²) had comparable plasma exposure of lumasiran and similar pharmacodynamics as patients with normal renal function (eGFR ≥90 mL/min/1.73 m²). In patients with moderate renal impairment (eGFR 30 to <60 mL/min/1.73 m²) Cmax was similar to that in patients with normal renal function; AUC was 25% higher based on limited data. Limited clinical data are available in patients with severe renal impairment (eGFR 15 to <30 mL/min/1.73 m²), end-stage renal disease (eGFR <15 mL/min/1.73 m²), or who are on dialysis (see section 4.2). For ESRD patients on dialysis, within the same body weight category, a transient 3- to 7-fold higher Cmax and 2- to 3.5-fold AUC0-last increase was observed (see section 5.2 Pharmacokinetic/pharmacodynamic relationship(s)). However, plasma concentrations decline below the level of detection within 24 to 48 hours, similar to patients without renal impairment.
Paediatric population
Data in children younger than 1 year of age are limited. In children <20 kg, lumasiran Cmax was 2-fold higher due to the nominally higher 6-mg/kg dose and faster absorption rate. The pharmacodynamics of lumasiran were comparable in paediatric patients (aged 4 months to 17 years) and in adults, despite the transiently higher plasma concentrations in children <20 kg, due to the rapid and predominant distribution of lumasiran to the liver.
Body weight
The recommended dosing regimens yielded up to 2-fold higher Cmax in children <20 kg while AUC was similar across the body weights studied (6.2 to 110 kg).
5.3. Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology and genotoxicity.
In rats, but not in monkeys, microscopic changes in the liver (e.g. hepatocellular vacuolation, mitosis and karyomegaly) were observed, accompanied by decrease in plasma fibrinogen levels and other laboratory changes. The reason for the apparent rodent-specificity is not understood and the relevance for humans is unclear.
Lumasiran did not show any adverse effects on male and female fertility and pre- and post-natal development in rats. In embryo-foetal development studies in rats and rabbits, skeletal abnormalities were observed, but at high exposure multiples relative to human therapeutic exposures. The NOAELs were approximately 20- to 70-times higher (based on monthly exposures).
A dose-range finding toxicity study in neonate rats did not show increased sensitivity of the developing rat to either the toxicology or pharmacology of lumasiran at exposure multiples of 2 compared to human therapeutic exposures (based on monthly exposures).
Animal studies have not been conducted to evaluate the carcinogenic potential of lumasiran.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.