PRAXBIND Solution for injection Ref.[8938] Active ingredients: Idarucizumab
Source: European Medicines Agency (EU) Revision Year: 2018 Publisher: Boehringer Ingelheim International GmbH, Binger Str. 173, D-55216, Ingelheim am Rhein, Germany
Pharmacodynamic properties
Pharmacotherapeutic group: all other therapeutic products, antidotes
ATC code: V03AB37
Mechanism of action
Idarucizumab is a specific reversal agent for dabigatran. It is a humanized monoclonal antibody fragment (Fab) that binds to dabigatran with very high affinity, approximately 300-fold more potent than the binding affinity of dabigatran for thrombin. The idarucizumab-dabigatran complex is characterised by a rapid on-rate and extremely slow off-rate resulting in a very stable complex. Idarucizumab potently and specifically binds to dabigatran and its metabolites and neutralises their anticoagulant effect.
Clinical efficacy and safety
Three randomised, double-blind, placebo-controlled Phase I studies in 283 subjects (224 treated with idarucizumab) were conducted to assess the safety, efficacy, tolerability, pharmacokinetics and pharmacodynamics of idarucizumab, given alone or after administration of dabigatran etexilate. The investigated population consisted of healthy subjects and subjects exhibiting specific population characteristics covering age, body weight, race, sex and renal impairment. In these studies the doses of idarucizumab ranged from 20 mg to 8 g and the infusion times ranged from 5 minutes to 1 hour.
Representative values for pharmacokinetic and pharmacodynamics parameters were established on the basis of healthy subjects aged 45-64 years receiving 5 g idarucizumab (see sections 5.1 and 5.2).
One prospective, open-label, non-randomized, uncontrolled study (RE-VERSE AD) was conducted to investigate the treatment of adult patients who presented with dabigatran-related life-threatening or uncontrolled bleeding (Group A) or who required emergency surgery or urgent procedures (Group B). The primary endpoint was the maximum percentage reversal of the anticoagulant effect of dabigatran within 4 hours after the administration of idarucizumab, based on central laboratory determination of dilute thrombin time (dTT) or ecarin clotting time (ECT). A key secondary endpoint was the restoration of haemostasis.
RE-VERSE AD included data for 503 patients: 301 patients with serious bleeding (Group A) and 202 patients requiring an urgent procedure/surgery (Group B). Approximately half of the patients in each group were male. The median age was 78 years and the median creatinine clearance was 52.6 mL/min. 61.5% of patients in Group A and 62.4% of patients in Group B had been treated with dabigatran 110 mg twice daily.
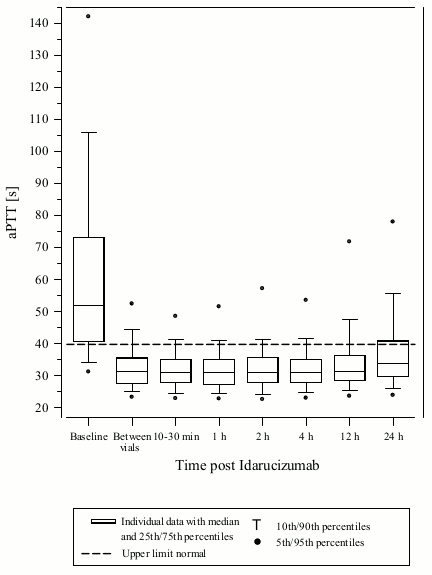
Reversal was only evaluable for those patients showing prolonged coagulation times prior to idarucizumab treatment. Most patients in both Groups A and B, achieved complete reversal of the anticoagulant effect of dabigatran (dTT: 98.7%; ECT: 82.2%; aPTT: 92.5% of evaluable patients, respectively) in the first 4 hours after administration of 5 g idarucizumab. Reversal effects were evident immediately after administration.
Figure 1. Reversal of dabigatran-induced clotting time prolongation determined by dTT in patients from the RE-VERSE AD study (N=487):
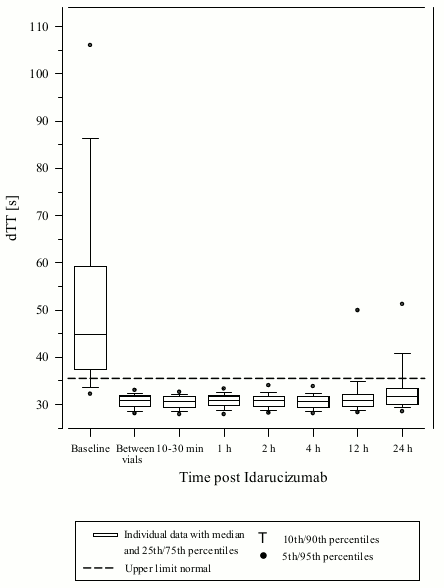
Figure 2. Reversal of dabigatran-induced clotting time prolongation determined by ECT in patients from the RE-VERSE AD study (N=487):
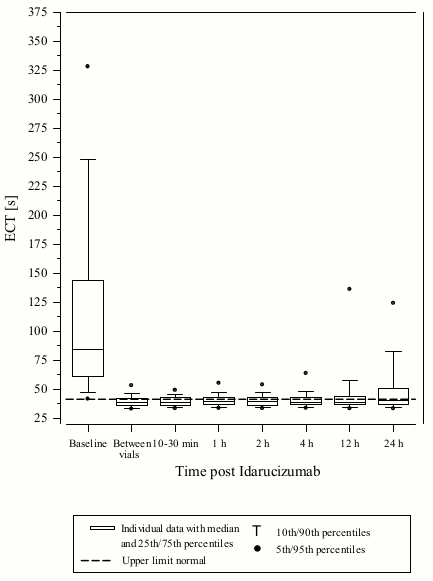
Figure 3. Reversal of dabigatran-induced clotting time prolongation determined by aPTT in patients from the RE-VERSE AD study (N=486):
Restoration of haemostasis was achieved in 80.3% of evaluable patients who had serious bleeding and normal haemostasis was observed in 93.4% of patients who required an urgent procedure.
Of the total 503 patients, 101 patients died; each of these deaths could be attributed either as a complication of the index event or associated with co-morbidities. Thrombotic events were reported in 34 patients (23 out of the 34 patients were not on antithrombotic therapy at the time of the event) and in each of these cases, the thrombotic event could be attributed to the underlying medical condition of the patient. Mild symptoms of potential hypersensitivity (pyrexia, bronchospasm, hyperventilation, rash or pruritus) were reported. A causal relationship to idarucizumab could not be established.
Pharmacodynamic effects
The pharmacodynamics of idarucizumab after administration of dabigatran etexilate were investigated in 141 subjects in Phase I studies, of which data for a representative subgroup of 6 healthy subjects aged 45 to 64 years receiving a dose of 5 g as intravenous infusion are presented. The median peak dabigatran exposure in the investigated healthy subjects was in the range of a twice daily administration of 150 mg dabigatran etexilate in patients.
Effect of idarucizumab on the exposure and anticoagulant activity of dabigatran
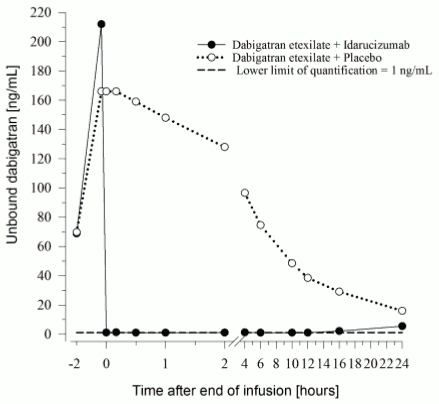
Immediately after the administration of idarucizumab, the plasma concentrations of unbound dabigatran were reduced by more than 99 %, resulting in levels with no anticoagulant activity.
The majority of the patients showed sustained reversal of dabigatran plasma concentrations up to 12 hours (≥90%). In a subset of patients, recurrence of plasma levels of unbound dabigatran and concomitant elevation of clotting tests was observed, possibly due to re-distribution of dabigatran from the periphery. This occurred 1-24 hours after administration of idarucizumab mainly at timepoints ≥12 hours.
Figure 4. Plasma-levels of unbound dabigatran in the representative group of healthy subjects (administration of idarucizumab or placebo at 0 h):
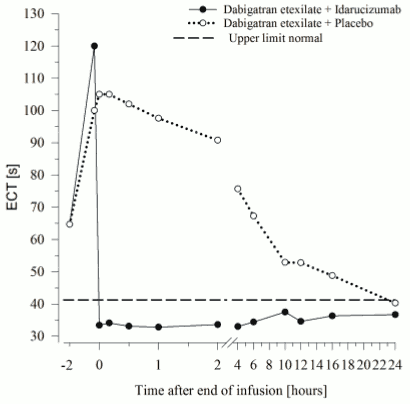
Dabigatran prolongs the clotting time of coagulation markers such as diluted Thrombin Time (dTT), Thrombin Time (TT), activated Partial Thromboplastin Time (aPTT) and Ecarin Clotting Time (ECT), which provide an approximate indication of the anticoagulation intensity. A value in the normal range after administration of idarucizumab indicates that a patient is no longer anticoagulated. A value above the normal range may reflect residual active dabigatran or other clinical conditions e.g., presence of other drugs or transfusion coagulopathy. These tests were used to assess the anticoagulant effect of dabigatran. A complete and sustained reversal of dabigatran-induced clotting time prolongation was observed immediately after the idarucizumab infusion, lasting over the entire observation period of at least 24 h.
Figure 5. Reversal of dabigatran-induced clotting time prolongation determined by dTT in the representative group of healthy subjects (administration of idarucizumab or placebo at 0 h):
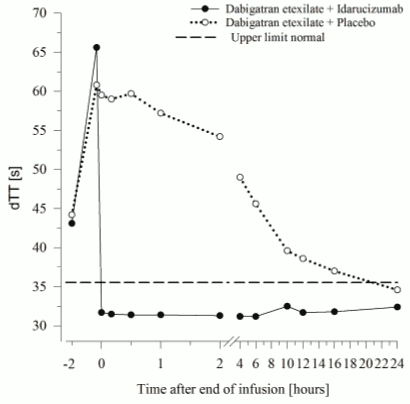
Figure 6. Reversal of dabigatran-induced clotting time prolongation determined by ECT in the representative group of healthy subjects (administration of idarucizumab or placebo at 0 h):
Thrombin generation parameters
Dabigatran exerts pronounced effects on parameters of the endogenous thrombin potential (ETP). Idarucizumab treatment normalised both thrombin lag time ratio and time to peak ratio to baseline levels as determined 0.5 to 12 hours after the end of the idarucizumab infusion. Idarucizumab alone has shown no procoagulant effect measured as ETP. This suggests that idarucizumab has no prothrombotic effect.
Re-administration of dabigatran etexilate
24 hours after the idarucizumab infusion, re-administration of dabigatran etexilate resulted in expected anticoagulant activity.
Immunogenicity
Serum samples from 283 subjects in phase I trials (224 volunteers treated with idarucizumab) and 501 patients were tested for antibodies to idarucizumab before and after treatment. Pre-existing antibodies with cross-reactivity to idarucizumab were detected in approximately 12 % (33/283) of the phase I subjects and 3.8% (19/501) of the patients. No impact on the pharmacokinetics or the reversal effect of idarucizumab or hypersensitivity reactions were observed.
Treatment-emergent possibly persistent anti-idarucizumab antibodies with low titers were observed in 4% (10/224) of the phase I subjects and 1.6% (8/501) of the patients suggesting a low immunogenic potential of idarucizumab. In a subgroup of 6 phase I subjects, idarucizumab was administered a second time, two months after the first administration. No anti-idarucizumab antibodies were detected in these subjects prior to the second administration. In one subject, treatment-emergent anti-idarucizumab antibodies were detected after the second administration. Nine patients were re-dosed with idarucizumab. All nine patients were re-dosed within 6 days after the first idarucizumab dose. None of the patients re-dosed with idarucizumab tested positive for anti-idarucizumab antibodies.
Preclinical pharmacodynamics
A trauma model in pigs was performed using a blunt liver injury after dosing with dabigatran to achieve supratherapeutic concentrations of about 10-fold of human plasma levels. Idarucizumab effectively and rapidly reversed the life-threatening bleeding within 15 min after the injection. All pigs survived at idarucizumab doses of approximately 2.5 and 5 g. Without idarucizumab, the mortality in the anticoagulated group was 100%.
The European Medicines Agency has deferred the obligation to submit the results of studies with Praxbind in one or more subsets of the paediatric population in the prevention and treatment of dabigatran associated haemorrhage (see section 4.2 for information on paediatric use).
Pharmacokinetic properties
The pharmacokinetics of idarucizumab were investigated in 224 subjects in Phase I studies, of which data for a representative subgroup of 6 healthy subjects aged 45 to 64 years receiving a dose of 5 g as intravenous infusion are presented.
Distribution
Idarucizumab exhibited multiphasic disposition kinetics and limited extravascular distribution. Following the intravenous infusion of a 5 g dose, the geometric mean volume of distribution at steady state (Vss) was 8.9 L (geometric coefficient of variation (gCV) 24.8%).
Biotransformation
Several pathways have been described that may contribute to the metabolism of antibodies. All of these pathways involve biodegradation of the antibody to smaller molecules, i.e. small peptides or amino acids, which are then reabsorbed and incorporated in the general protein synthesis.
Elimination
Idarucizumab was rapidly eliminated with a total clearance of 47.0 mL/min (gCV 18.4%), an initial half-life of 47 minutes (gCV 11.4%) and a terminal half-life of 10.3 h (gCV 18.9%). After intravenous administration of 5 g idarucizumab, 32.1% (gCV 60.0%) of the dose was recovered in urine within a collection period of 6 hours and less than 1% in the following 18 hours. The remaining part of the dose is assumed to be eliminated via protein catabolism, mainly in the kidney.
After treatment with idarucizumab proteinuria has been observed. The transient proteinuria is a physiologic reaction to renal protein overflow after bolus/short term application of 5g idarucizumab intravenously. The transient proteinuria usually peaked about 4 h after idarucizumab administration and normalised within 12-24 hours. In single cases the transient proteinuria persisted for more than 24 hours.
Patients with renal impairment
In Phase I studies Praxbind has been investigated in subjects with a creatinine clearance ranging from 44 to 213 mL/min. Subjects with a creatinine clearance below 44 mL/min have not been studied in Phase I. Depending on the degree of renal impairment the total clearance was reduced compared to healthy subjects, leading to an increased exposure of idarucizumab.
Based on pharmacokinetic data from 347 patients with different degrees of renal function (median creatinine clearance 21-99 mL/min) it is estimated that mean idarucizumab exposure (AUC0–24h) increases by 38% in patients with mild (CrCl 50-<80 mL/min), by 90% in moderate (30-<50 mL/min) and by 146% in severe (0-<30 mL/min) renal impairment. Since dabigatran is also excreted primarily via the kidneys, increases in the exposure to dabigatran are also seen with worsening renal function.
Based on these data and the extent of reversal of the anticoagulant effect of dabigatran in patients, renal impairment does not impact the reversal effect of idarucizumab.
Patients with hepatic impairment
An impact of hepatic impairment, assessed by hepatic injury as determined by elevated liver function tests, on the pharmacokinetics of idarucizumab has not been observed.
Idarucizumab has been studied in 58 patients with varying degrees of hepatic impairment. Compared to 272 patients without hepatic impairment, the median AUC of idarucizumab was changed by -6%, 37% and 10% in patients with AST/ALT elevations of 1 to <2x ULN (N=34), 2 to <3x ULN (N=3) and >3x ULN (N=21), respectively. Based on pharmacokinetic data from 12 patients with liver disease, the AUC of idarucizumab was increased by 10% as compared to patients without liver disease.
Older people/Sex/Race
Based on population pharmacokinetic analyses, sex, age, and race do not have a clinically meaningful effect on the pharmacokinetics of idarucizumab.
Preclinical safety data
Preclinical data reveal no special hazard for humans based on repeated dose toxicity studies of up to four weeks in rats and two weeks in monkeys. Safety pharmacology studies have demonstrated no effects on the respiratory, central nervous or cardiovascular system.
Studies to evaluate the mutagenic and carcinogenic potential of idarucizumab have not been performed. Based on its mechanism of action and the characteristics of proteins no carcinogenic or genotoxic effects are anticipated.
Studies to assess the potential reproductive effects of idarucizumab have not been performed. No treatment-related effects have been identified in reproductive tissues of either sex during repeat dose intravenous toxicity studies of up to four weeks in the rat and two weeks in monkeys. Additionally, no idarucizumab binding to human reproductive tissues was observed in a tissue cross-reactivity study. Therefore, preclinical results do not suggest a risk to fertility or embryo-fetal development.
No local irritation of the blood vessel was observed after i.v. or paravenous administration of idarucizumab. The idarucizumab formulation did not produce haemolysis of human whole blood in vitro.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.