Source: European Medicines Agency (EU) Revision Year: 2015 Publisher: Dendreon UK Limited, 41 Chalton Street, London, NW1 1JD, United Kingdom Tel: (0)20 7554 2222 Fax: (0)20 7554 2201 dendreonuk@dendreon.com
Pharmacotherapeutic group: Immunostimulants, other immunostimulants
ATC code: L03AX17
Provenge is an autologous cellular immunotherapy designed to induce an immune response targeted against prostatic acid phosphatase (PAP), an antigen expressed in most prostate cancers. Peripheral blood mononuclear cells collected from the patients are cultured with PAP-GM-CSF, a fusion protein consisting of PAP linked to granulocyte-macrophage colony-stimulating factor (GM-CSF) an immune cell activator. During ex vivo culture with PAP-GM-CSF, activated APCs (antigen presenting cells) take up and process the recombinant target antigen into peptides that are then presented to T cells. Product characterization shows that PAP and PAP-GM-CSF fusion protein-specific T cells are generated during treatment and are detected in the peripheral blood of patients after treatment with Provenge.
As part of lot release, each product is assessed for activation of antigen presenting cells (APCs) by virtue of increased surface CD54 expression after culture with PAP-GM-CSF. CD54 is an adhesion and costimulatory molecule essential in the formation of the immunological synapse between an APC and a T cell. The degree of CD54 upregulation correlates with overall survival in the randomised controlled clinical trials carried out with Provenge in metastatic castrate resistant prostate cancer. In clinical study D9902B (IMPACT), 237 out of the 512 patients randomized were evaluated for the development of humoral or cellular immune responses (T cell proliferation and gamma-interferon (γIFN) ELISPOT) to the target antigens at baseline, and at Weeks 6, 14, and 26. Antibody (IgM and IgG) responses against both PAP-GM-CSF and the PAP antigens were observed in the Provenge group through the follow up period. T cell proliferative and γIFN ELISPOT responses to PAP and PAP-GM-CSF were observed in cells collected from peripheral blood of patients through the follow-up period in the Provenge treatment group but not in controls. There was a correlation between cellular or antibody responses to PAP or PAP-GM-CSF in the Provenge group and improved survival. Neutralising antibody responses to GM-CSF were transient.
The efficacy and safety of Provenge in patients with asymptomatic or minimally symptomatic metastatic castrate resistant prostate cancer were studied in three similar Phase III, randomised, double-blind, controlled, multicentre trials: D9902B (IMPACT), D9901, and D9902A. The patients enrolled in these trials had failed surgical or medical castration (e.g. luteinising hormone-releasing hormone [LHRH] agonist or gonadotropin-releasing hormone [GnRh] antagonist) therapies and had metastatic disease in the soft tissue and/or bone. Patients did not require opioid analgesics for pain management and the majority had not received prior chemotherapy.
Following randomization, patients from both treatment groups underwent a series of 3 leukapheresis procedures (at approximately 2 week intervals, range 1 to 15 weeks). Each leukapheresis was followed approximately 3 days later by infusion of Provenge or control. The control was nonactivated autologous peripheral blood mononuclear cells. Following disease progression, patients were treated at the physician’s discretion with other anti-cancer interventions. Patients in the control group could enrol in an open-label protocol and receive an investigational autologous cellular therapy manufactured from cells cryopreserved at the time the control product was prepared.
The IMPACT study was a randomised, double-blind, controlled, multicentre trial in patients with asymptomatic or minimally symptomatic metastatic castrate resistant prostate cancer. Eligible patients had metastatic disease in the soft tissue and/or bone with current or historical evidence of disease progression concomitant with surgical or medical castration, as evidenced by progression of serum prostate specific antigen (PSA) and/or bone or soft tissue disease, and an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1. Exclusion criteria included visceral (liver, lung, or brain) metastases, malignant pleural effusions or malignant ascites, pathologic long bone fractures, imminent pathologic long-bone fractures (cortical erosion on radiography >50%), spinal cord compression, moderate to severe prostate cancer-related pain and use of narcotics for cancerrelated pain, and treatment with chemotherapy at least 3 months prior to randomisation. The primary endpoint was overall survival. Secondary endpoints included time to objective disease progression, time to clinical progression, and PSA doubling time (PSADT).
A total of 512 patients were randomised in a 2:1 ratio to receive Provenge (n=341) or control (n=171). The median age was 71, 90% of the patients were Caucasian, and all had a life expectancy of at least 6 months. Thirty-five percent of patients had undergone radical prostatectomy, 54% had received local radiotherapy, and 82% had received combined androgen blockade. All patients had baseline testosterone levels <50 ng/mL. Forty-eight percent of patients were receiving bisphosphonates and 18% had received prior chemotherapy, including docetaxel. Eighty-two percent of patients had an ECOG performance status of 0; 75% had a Gleason sum ≤7; 44% had bone and soft tissue disease; 48% had bone-only disease; 7% had soft tissue-only disease; and 43% had greater than ten bony metastases.
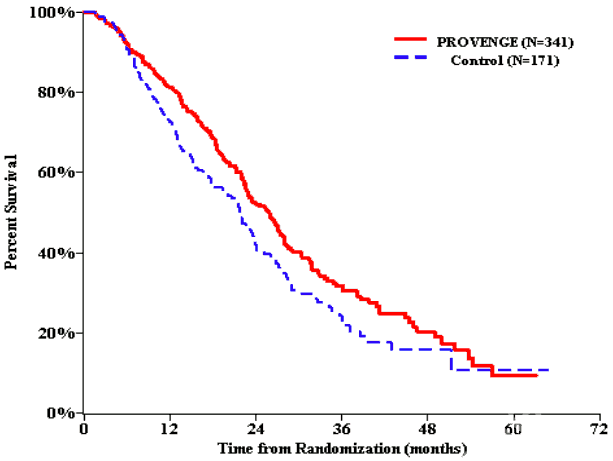
A statistically significant improvement in overall survival was seen in patients treated with Provenge, with a 22.5% reduction in the risk of death compared with control (see Table 2 and Figure 1). Of the control arm patients, 64% crossed over to receive an investigational autologous cellular immunotherapy manufactured from cells cryopreserved at the time the control was manufactured; patients were not randomised to subsequent autologous cellular immunotherapy.
Figure 1. Kaplan-Meier overall survival curve, IMPACT study:
A retrospective subgroup analysis has suggested a greater Provenge treatment effect in patients with a baseline PSA <22.1 ng/mL [HR= 0.521 (95% CI: 0.309, 0.879)]. Intermediate results were observed in patients with baseline PSA >22.1 to 50.1 ng/mL [HR=0.685 (95% CI: 0.431, 1.088)] and patients with baseline PSA >50.1 to 134.1 ng/mL [HR=0.819 (95% CI: 0.532, 1.262)]. A smaller treatment effect was observed in those with baseline PSA >134.1 ng/mL [HR=0.853 (95% CI: 0.554, 1.315)].
Analyses of time to objective disease progression, time to clinical progression, or PSA doubling time (PSADT) did not meet statistical significance.
Study D9901 was a randomised, double-blind, controlled, multicentre trial in patients with metastatic castrate resistant prostate cancer and no cancer-related pain. The primary endpoint was time to disease progression, which did not reach statistical significance. Overall survival was not a study endpoint but a pre-specified analysis. Patients treated with Provenge had a statistically significant survival advantage compared with control.
A third study, D9902A, similar in design to study D9901, was terminated prior to completion of planned accrual based on the time to disease progression results in study D9901. The primary endpoint was time to disease progression and the secondary endpoint was overall survival. Neither endpoint met statistical significance.
Table 2 presents overall survival results observed in IMPACT, study D9901, and study D9902A.
Table 2. Summary of overall survival (all patients as randomized):
| IMPACT | D9901 | D9902A | ||||
|---|---|---|---|---|---|---|
| Provenge (N=341) | Control (N=171) | Provenge (N=82) | Control (N=45) | Provenge (N=65) | Control (N=33) | |
| Overall survival Median, months (95% CI) | 25.8 (22.8, 27.7) | 21.7 (17.7, 23.8) | 25.9 (20.0, 32.4) | 21.4 (12.3, 25.8) | 19.0 (13.6, 31.9) | 15.7 (12.8, 25.4) |
| Hazard ratio (95% CI) | 0.775a (0.614, 0.979) | 0.586b (0.388, 0.884) | 0.786b (0.484, 1.278) | |||
| p-value | 0.032a | 0.010c | 0.331c | |||
| 36-month survival (%) | 32% | 23% | 34% | 11% | 32% | 21% |
a Hazard ratio and p-value based on the Cox Model adjusted for PSA (ln) and LDH (ln) and stratified by bisphosphonate use, number of bone metastases, and primary Gleason grade.
b Hazard ratio based on the unadjusted Cox Model.
c p-value based on a log-rank test.
Abbreviations: CI = confidence interval.
The European Medicines Agency has waived the obligation to submit the results of studies with Provenge in all subsets of the paediatric population in the treatment of prostate cancer (see section 4.2 for information on paediatric use).
Provenge is an autologous cellular therapy. The nature of Provenge is such that conventional studies on pharmacokinetics, absorption, distribution, metabolism, and elimination are not applicable.
Conventional toxicology, carcinogenicity, mutagenicity, and reproductive toxicity studies have not been performed.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.