REDEMPLO Solution for injection Ref.[116717] Active ingredients: Plozasiran
Source: European Medicines Agency (EU) Revision Year: 2026 Publisher: Arrowhead Pharmaceuticals Ireland Limited, One Spencer Dock, North Wall Quay, Dublin 1, D01 X9R7, Ireland
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Lipid modifying agents, other lipid modifying agents
ATC code: not yet assigned
Mechanism of action
Plozasiran is a small interfering RNA (siRNA, double-stranded oligonucleotide) conjugated with N-acetylgalactosamine to facilitate delivery to and uptake by hepatocytes. In hepatocytes, plozasiran selectively degrades the mRNA for apolipoprotein C3 (APOC3) through the RNA interference mechanism resulting in reduced levels of hepatic and serum APOC3 protein. This, in turn, enhances the activity of lipoprotein lipase and hepatocyte uptake of TG-rich lipoprotein remnants leading to decreases in serum TG.
Pharmacodynamic effects
In the PALISADE study, 25 mg plozasiran administered every 3 months in patients with FCS decreased APOC3, TG, non-high density lipoprotein cholesterol (non-HDL-C), and very low-density lipoprotein cholesterol (VLDL-C) (see also below under "Clinical efficacy") and increased HDL-C and LDL-C. LDL-C levels remained within the normal range for most patients. The median reductions in fasting serum APOC3 protein and TG at month 1 were 95% and 85%, respectively, suggesting pharmacodynamic steady-state is achieved following the first dose.
Cardiac electrophysiology
Doses of 100 mg plozasiran (4 times the recommended dose) did not prolong the QT interval to any clinically relevant extent.
Clinical efficacy
PALISADE study in patients with FCS
PALISADE is a randomised, double-blind, placebo-controlled clinical study in 75 adult patients with FCS maintained on a low-fat diet. Patients ≥18 years of age received 4 single subcutaneous injections of either 25 mg plozasiran (N=23), 50 mg plozasiran (N=22) or placebo (N=19) administered every 3 months. Patients with a diagnosis of FCS and fasting TGs ≥10 mmol/L (≥880 mg/dL) that were refractory to standard lipid-lowering therapy were included.
A diagnosis of FCS was defined as patients with a history of fasting TGs >11.3 mmol/L (˃1,000 mg/dL) and either:
- A supportive genetic test (N=41 [54.7%]) or evidence of low lipoprotein lipase (LPL) activity; or
- Clinically diagnosed FCS (N=34 [45.3%]) with either prior history of acute pancreatitis not caused by alcohol or cholelithiasis, history of recurrent hospitalisations for severe abdominal pain without other explainable cause, history of childhood pancreatitis, or family history of hypertriglyceridaemia-induced pancreatitis.
The mean age was 46 years with more patients in the 50 mg plozasiran group being <50 years of age (83.3%) than in the 25 mg plozasiran or placebo groups (57.7% and 56.0%, respectively). The number of patients ≥65 years of age was 9 (12%) and those ≥75 years of age was 2 (3%). Approximately half of the patients in each treatment group were male. Most patients were White (73.3%) or Asian (21.3%). Mean body mass index (BMI) was 25.5 kg/m²; 53.3% of subjects were overweight (BMI ≥25 kg/m²). The number of patients with genetically confirmed FCS was 41, with 34 patients without genetic confirmation of FCS. Of the patients who received plozasiran, five variants were represented: APOA5 – 2.3%, APOC2 - 2.3%, GPIHBP1 – 9.1%, LMF1 – 6.8%, LPL – 81.8%. A total of 89.3% of the patients had experienced a prior episode of pancreatitis. Percentages of patients on TG lowering therapies at baseline were as follows: 66.7% were on fibrates, 29.3% were on icosapent ethyl, omega-3 fatty acid or fish oil, and 45.3% were on statins.
The majority of patients received all 4 planned doses; 24 (92.3%) patients in the 25 mg plozasiran group, 22 (91.7%) patients in the 50 mg plozasiran group and 19 (76.0%) patients in the placebo group.
The primary efficacy endpoint was median percent change from baseline at month 10 in fasting TGs. At month 10, plozasiran statistically significantly reduced median fasting TG levels at the 25 mg recommended dose (see Table 2). The TG lowering effects of 50 mg plozasiran did not offer a therapeutic benefit over the recommended 25 mg dose.
In the PALISADE study, 25 mg plozasiran administered every 3 months in patients with FCS significantly reduced median fasting serum APOC3 protein by 93% (p<0.0001).
The reductions in TG levels observed in plozasiran-treated patients were apparent at month 1 (first post-baseline measurement) and remained consistent throughout the 12‑month duration of the PALISADE study with relatively small peak-to-trough fluctuations (see Figure 1). Median TG levels achieved at several timepoints throughout the treatment period were below the recognised threshold of 5.7 mmol/L (500 mg/dL) for increased risk of acute pancreatitis (see Figure 1).
Table 2. Median difference in percent change from baseline in fasting TG and APOC3 in patients with FCS at month 10 in PALISADE study:
| Treatment group | Placebo | Plozasiran 25 mg |
| Baseline TG (mmol/L) | ||
| N | 25 | 26 |
| Median | 23.2 | 22.7 |
| Month 10 TG (mmol/L) | ||
| N | 19 | 24 |
| Median | 18.2 | 5.0 |
| Median percent change at month 10 from baseline in fasting TG | -17 | -80 |
| Difference from placebo | -58.7 | |
| 95% CI | -89.6, -27.9 | |
| p-value | p<0.0001 | |
| Median percent change at month 10 from baseline in fasting APOC3 | -1.3 | -93.0 |
| Difference from placebo | -90.5 | |
| 95% CI | -108.3, -72.7 | |
| p-value | p<0.0001 | |
APOC3 = apolipoprotein C3; CI = confidence interval; FCS = familial chylomicronaemia syndrome;
TG = triglyceride.
Figure 1. Median absolute fasting triglyceride levels in patients with FCS during the PALISADE study:
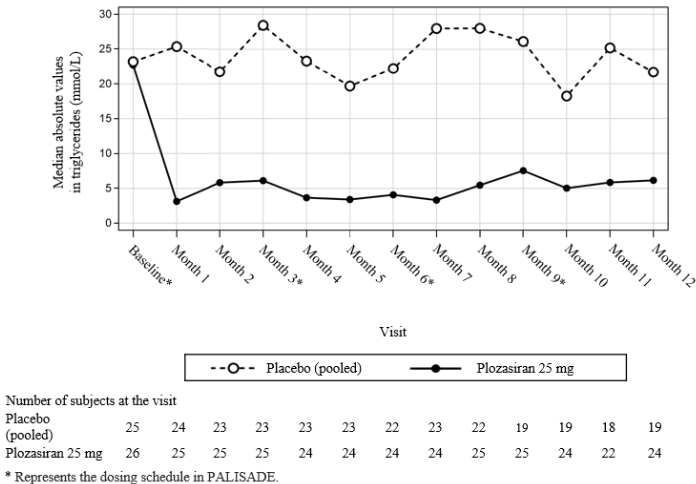
A prespecified subgroup analysis of genetically confirmed versus clinically diagnosed FCS patients showed that patients had a similar TG response to plozasiran independent of their confirmed genetic characteristics.
Among patients with fasting TG measurements at month 10, all patients in the 25 mg plozasiran group experienced decreases from baseline and approximately 80% of patients experienced at least a >50% decrease from baseline. In addition, when compared to placebo, the combined doses of 25 mg and 50 mg plozasiran significantly reduced the incidence of acute pancreatitis (odds ratio, 0.169; p=0.0292). The odds of acute pancreatitis were 83% lower in the pooled plozasiran groups compared with the placebo group, with 7 pancreatitis events occurring in 5 (20%) patients in the placebo group and 2 pancreatitis events occurring in 2 (4%) patients in the pooled plozasiran groups.
PALISADE open label extension (OLE) study in patients with FCS
Of the 64 patients who completed 12 months of randomised study treatment, 62 (97%) entered the OLE period. Of these patients, 18 (29%) received placebo (placebo/plozasiran group) and 44 (71%) received plozasiran (plozasiran/plozasiran group) during the randomised period.
As expected, median absolute values of fasting TGs at OLE baseline (month 12) were higher in patients who received placebo in the randomised period (placebo/plozasiran group; 23.76 mmol/L [2 103 mg/dL]) compared to the plozasiran/plozasiran group (6.31 mmol/L [558 mg/dL]). Notably, for those in the placebo/plozasiran group, median TGs had already fallen to a level similar to the plozasiran/plozasiran group after the first month of plozasiran treatment (month 13; 3.67 mmol/L [325 mg/dL; -87.96%] and 6.0 mmol/L [531 mg/dL; -75.23%] in the placebo/plozasiran and plozasiran/plozasiran groups, respectively); allowing for the expected variability in fasting TGs and measurements taken at trough, these reductions were sustained through month 18 of the OLE period.
Immunogenicity
In the PALISADE study, none of the 50 FCS patients treated with plozasiran over a period of 12 months developed treatment-induced or treatment-boosted anti-drug antibodies (ADA). There was no evidence to indicate that plozasiran pharmacodynamics or efficacy changed over time following multiple administrations of plozasiran. No adverse effects related to systemic immunoreaction were found in the plozasiran-treated patients.
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with plozasiran in one or more subsets of the paediatric population in the treatment of familial chylomicronaemia syndrome (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Absorption
Following a single subcutaneous injection of 25 mg plozasiran, the peak plasma concentration (Cmax) was 68.5 ng/mL. The median time to reach Cmax (Tmax) was 6 hours.
Plozasiran has not been administered intravenously in any clinical studies, therefore, absolute bioavailability data in humans are not available. Following subcutaneous administration in cynomolgus monkeys, the absolute bioavailability of plozasiran was estimated to be 40%.
Distribution
Following repeated subcutaneous injections of 25 mg plozasiran, it is distributed in plasma and extracellular body water with apparent volume of distribution (Vz/F) of 146 L in the terminal-phase of elimination. Once in systemic circulation, plozasiran is primarily distributed to the liver. In plasma, plozasiran has an unbound fraction of 22%.
In vitro studies suggest that plozasiran is not a substrate, inhibitor, or inducer of transporters. Therefore, plozasiran is not expected to cause or be affected by interactions mediated through transporters.
Biotransformation
Plozasiran is primarily metabolised by nucleases in the liver to shorter oligonucleotides of varying lengths. In vitro studies suggest that plozasiran is not a substrate of cytochrome P450 (CYP450) enzymes.
In vitro studies suggest that plozasiran is not a substrate, inhibitor, or inducer of CYP450 enzymes. Therefore, plozasiran is not expected to cause or be affected by interactions mediated through CYP450 enzymes.
Elimination
The terminal elimination half-life of plozasiran in plasma is approximately 3–4 hours. The mean apparent systemic clearance is 33.8 L/hour. Approximately 16–19% of plozasiran dose is excreted in the urine.
Linearity/non-linearity
Plozasiran exhibited time-invariant pharmacokinetics following repeated subcutaneous injections. Following multiple dose administrations, plasma levels of plozasiran (Cmax, AUC0-t and AUC0-inf) increased proportionally with dose within the dose range of 10–50 mg.
Pharmacokinetic/pharmacodynamic relationship(s)
Plozasiran is active inside hepatocytes with prolonged pharmacodynamic activity that is disconnected from its pharmacokinetic profile in the plasma compartment. The long duration of action is beyond the plasma elimination half-life of 3–4 hours. Pharmacodynamic response is likely saturated at the recommended dose of 25 mg plozasiran every 3 months.
Immunogenicity
In the PALISADE study, none of the 50 FCS patients treated with plozasiran over a period of 12 months developed treatment-induced or treatment-boosted anti-drug antibodies (ADA). There was no evidence to indicate that plozasiran pharmacokinetics changed over time following multiple administrations of plozasiran.
Special populations
Elderly
No clinically significant differences in plozasiran pharmacokinetics based on age were found in a population pharmacokinetic analysis conducted with data from adult healthy subjects and patients (N=146); age 65–74 years (N=16); age 75–85 years (N=4) (see section 4.2).
Renal impairment
No clinically significant differences in plozasiran pharmacokinetics based on mild (eGFR ≥60 to ˂90 mL/min) or moderate (eGFR ≥30 to ˂60 mL/min) renal impairment were found in a population pharmacokinetic analysis that included data from 23 and 4 patients with mild and moderate degrees of renal impairment, respectively. Plozasiran has not been studied in patients with severe renal impairment or end-stage renal disease (eGFR ˂30 mL/min) (see section 4.2).
Hepatic impairment
No clinically significant differences in plozasiran pharmacokinetics were found in a population pharmacokinetic analysis from 4 patients with elevation of AST > ULN and total bilirubin ≤ ULN, or total bilirubin >1.0 to 1.5 × ULN and any AST. Plozasiran has not been studied in patients with moderate or severe hepatic impairment (see section 4.2).
Body weight, BMI
Plozasiran plasma exposures (Cmax and AUC) are typically lower in patients with higher body weight or BMI without reduced treatment efficacy, and therefore no dose adjustment is recommended for heavier patients.
Gender, race, ethnicity
No clinically significant differences in plozasiran pharmacokinetics based on gender and race or ethnicity were found in a population pharmacokinetic analysis that included data from 65 (44.5%) females and 81 (55.5%) males with diverse race or ethnicity (67.1% White, 11.0% Black, 9.6% Asian, 2.1% Native Hawaiian or Pacific Islander, and 10.3% multiracial or unknown).
5.3. Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential, and toxicity to reproduction and development.
In a pre- and post-natal development study, there was an increase in the number of stillborn pups and a subsequent reduction in live birth index at the high dose, with a body surface area (BSA) adjusted safety margin of 3.1- and 31-fold at the preweaning and maternal/postnatal no observed adverse effect level (NOAEL).
There is no information on the excretion of plozasiran or its metabolites in animal milk.
In a 2-year rat carcinogenicity study, benign hepatocellular adenomas and a low incidence of carcinomas were noted at the high dose. Safety margins at the NOAEL are 10- and 16-fold based on BSA, and 60- and 53-fold based on AUC for males and females, respectively. Although the relevance for humans is unknown, the risk is likely low due to the high safety margins.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.