REVTORPYK Powder for solution for injection Ref.[116743] Active ingredients: Gedatolisib
Source: FDA, National Drug Code (US) Revision Year: 2026
12.1. Mechanism of Action
Gedatolisib is a kinase inhibitor of class I PI3K isoforms (α, β, δ, γ) and mTOR complexes mTORC1 and mTORC2, resulting in downstream inhibition of multiple effectors, including AKT. In estrogen receptor (ER)-positive breast cancer cell lines and xenograft models harboring wild-type or mutant PIK3CA, gedatolisib induced apoptosis and anti-proliferative effects in vitro and reduced tumor cell growth in vivo. In a human ER-positive breast cancer xenograft model harboring a PIK3CA mutation, the combination of gedatolisib with palbociclib and fulvestrant increased tumor growth inhibition compared to each treatment alone or the doublet combinations.
12.2. Pharmacodynamics
Exposure-Response Relationships
The exposure-response relationship and time course of pharmacodynamic response for the effectiveness of gedatolisib have not been fully characterized. An increased incidence of stomatitis or mucositis (Grade 3 and 4), nausea, vomiting, hyperglycemia, and adverse reaction leading to dosage modification or discontinuation was observed with higher gedatolisib exposure at the dose range of 10 mg to 319 mg (0.06 to 1.8 times the approved recommended dose).
Cardiac Electrophysiology
At weekly doses up to 319 mg (1.8 times the recommended dose), clinically significant QTc interval prolongation was not observed.
12.3. Pharmacokinetics
Gedatolisib pharmacokinetic parameters were observed at the approved recommended dosage in patients with breast cancer and are presented as mean (% coefficient of variation [%CV]), unless otherwise specified. Gedatolisib maximum plasma concentration (Cmax) is 13,700 (29%) ng/mL and area under the curve (AUC) is 21,400 (21%) ng·h/mL. Gedatolisib Cmax and AUC values increase dose proportionally in the dose range 10 mg to 319 mg (0.06 to 1.8 times the recommended dose). No accumulation of gedatolisib is observed after once-weekly administration.
Distribution
Gedatolisib plasma protein binding is 98%, and is not concentration dependent, in vitro. Gedatolisib blood-to- plasma ratio is 0.87, in vitro. Gedatolisib volume of distribution is 393 L.
Elimination
Gedatolisib elimination half-life (t1/2) is 36 hours with an estimated clearance of 8 (23%) L/hour.
Excretion
After a single dose of radiolabeled gedatolisib 89 mg to healthy subjects, approximately 67.4% of the dose was recovered in feces (67% unchanged) and 12.2% in urine (11.6% unchanged).
Specific Populations
No clinically significant effects on the pharmacokinetics of gedatolisib were observed based on age (21 to 84 years), sex, race (White [78%], Asian [9.1%], Black or African American [4.1%]) body weight (35 to 168 kg), mild hepatic impairment (total bilirubin ≤1.5 × ULN and any AST) or estimated glomerular filtration rate (eGFR) ≥45 mL/min/1.73 m² (MDRD equation). The effect of moderate (total bilirubin >1.5 to 3 times ULN with any AST) or severe (total bilirubin >3 x ULN with any AST) hepatic impairment, eGFR <45 mL/min/1.73 m², or dialysis on gedatolisib pharmacokinetics is unknown.
Drug Interactions
Clinical Studies and Model-Informed Approaches
No clinically significant differences in the pharmacokinetics of the following drugs were observed when used concomitantly with gedatolisib: metformin (MATE1, MATE2-K, and OCT1 substrate), rosuvastatin (BCRP substrate), or repaglinide (CYP2C8 sensitive substrate).
In vitro Studies
CYP450 Enzymes: Gedatolisib is not an inhibitor of CYP1A2, CYP2A6, or CYP2B6.
The ability of gedatolisib to induce CYP450 enzymes is unknown given the results of the vitro studies were inconclusive due to decreased viability of hepatocytes.
Transporter Systems: Gedatolisib is a substrate of P-gp and BCRP and is not a substrate for OATP1B1, OATP1B3, OCT1, NTCP, MRP2, or MRP3.
Gedatolisib did not inhibit P-gp, OATP1B1, OATP1B3, OAT1, OAT3, OCT2, and BSEP.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Carcinogenicity studies have not been conducted with gedatolisib.
Mutagenesis
Gedatolisib was not mutagenic in vitro in a bacterial reverse mutation (Ames) assay and was not clastogenic in an in vitro human lymphocyte chromosomal aberration assay. Gedatolisib was not genotoxic in an in vivo rat bone marrow micronucleus assay.
Impairment of Fertility
Fertility studies with gedatolisib have not been conducted. In repeated-dose toxicity studies, gedatolisib was administered intravenously once weekly on a 3 weeks on/1 week off schedule for 3 months in rats and dogs. Degeneration/atrophy of seminiferous tubules in testes was observed in male rats and dogs at ≥5 mg/kg and 0.5 mg/kg, respectively, and atrophy of prostate was observed in male rats at ≥1 mg/kg. In female rats, atrophy in the vagina and uterus was observed at ≥5 mg/kg. All findings occurred at exposures ≤0.2 times the human AUC at the recommended dose. Findings in males and females were reversible following a 12-week recovery period.
14. Clinical Studies
The efficacy of REVTORPYK in combination with fulvestrant, with or without palbociclib, was evaluated in Study 1 of VIKTORIA-1 (NCT05501886), an open label, randomized, multicenter clinical trial that enrolled 392 adult patients with locally advanced (inoperable) or metastatic HR-positive, HER2-negative (defined as immunohistochemistry (IHC) 0 or 1+, or IHC 2+/in situ hybridization (ISH-)) breast cancer without a detectable PIK3CA mutation. The PIK3CA status was assessed centrally for the following PIK3CA mutations: C420R, E542K, E545A, E545D [1635G>T only], E545G, E545K, Q546E, H1047L, H1047R, and H1047Y.
All patients were required to have progression on or after treatment with a CDK4/6 inhibitor and a non-steroidal aromatase inhibitor (AI) therapy. Patients could have received up to two prior lines of endocrine therapy for locally advanced (inoperable) or metastatic disease. Patients were excluded from the trial if they had Type 1 or uncontrolled Type 2 diabetes mellitus (requiring systemic insulin therapy), HbA1c >6.4%, and prior treatment with a PI3K inhibitor, AKT inhibitor, or mTOR inhibitor.
Patients were randomized (1:1:1) to three arms as follows:
- Arm A (N=131): REVTORPYK 180 mg intravenously once weekly for 3 weeks on followed by 1 week off (Days 1, 8, 15 for each 28-day cycle) in combination with fulvestrant 500 mg intramuscularly on cycle 1 Days 1 and 15, and then at Day 1 of each subsequent 28-day cycle, and palbociclib 125 mg orally once daily for 21 days followed by 7 days off treatment for each 28-day cycle (G+F+P), or
- Arm B (N=130): REVTORPYK 180 mg intravenously once weekly for 3 weeks on followed by 1 week off (Days 1, 8, 15 for each 28-day cycle) in combination with fulvestrant 500 mg intramuscularly on cycle 1 Days 1 and 15, and then at Day 1 of each subsequent 28-day cycle (G+F), or
- Arm C (N=131): Fulvestrant 500 mg intramuscularly on cycle 1 Days 1 and 15, and then at Day 1 of each subsequent 28-day cycle (F).
In addition, all pre/perimenopausal women were required to receive a gonadotropin-releasing hormone (GnRH) agonist. Randomization was stratified by presence of visceral metastases (yes or no), duration on immediate prior therapy (≤6 months or >6 months) and geographical region (Region 1: US or Region 2: Rest of World). Patients received treatment until disease progression or unacceptable toxicity. Patients on Arm C had the option to cross over to study Arm A or Arm B at the time of disease progression.
The major efficacy outcome was comparison of progression-free survival (PFS) assessed by blinded independent central review between patients enrolled in Arm A (G+F+P) and Arm C (F), and between patients enrolled in Arm B (G+F) and Arm C (F) evaluated according to Response Evaluation Criteria in Solid Tumors (RECIST), version 1.1. Additional efficacy outcome measures were overall survival (OS), objective response rate (ORR) and duration of response (DoR).
The baseline demographic and disease characteristics were: median age 56 years (range: 28 to 83 years); 99% female, of which 26% were pre/perimenopausal; 70% White, 16% Asian, 7% unknown, 2% Black or African American; 65% Not Hispanic/Latino, 28% Hispanic or Latino, 7% not reported/unknown; 100% had Stage IV breast cancer; all patients had prior endocrine therapy and prior CDK4/6 inhibitor therapy; 79% had visceral metastases; and Eastern Cooperative Oncology Group (ECOG) performance status of 0 (59%) or 1 (41%).
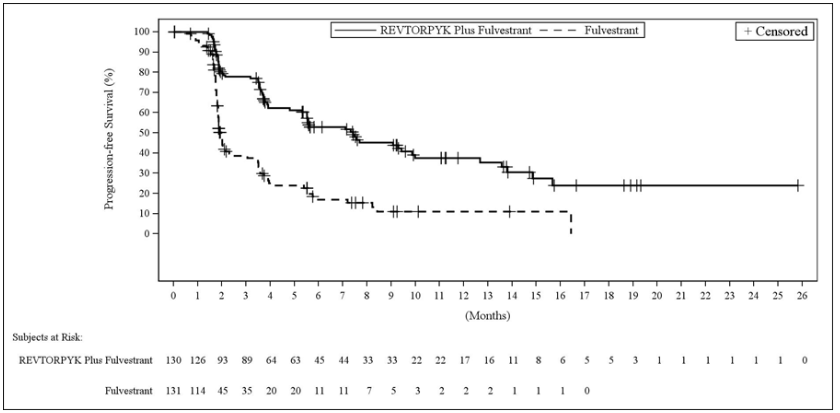
Efficacy results are summarized in Table 6 and Figures 1 and 2. At the time of the PFS analysis, OS data were not mature with 25% deaths in the overall population.
Table 6. Efficacy Results in Patients with Locally Advanced or Metastatic Breast Cancer in Study 1 of VIKTORIA-1:
| REVTORPYK + Fulvestrant + Palbociclib N=131 | REVTORPYK + Fulvestrant N=130 | Fulvestrant N=131 | |
| Progression-Free Survival (PFS) | |||
| Patients with events, n (%) | 59 (45) | 69 (53) | 89 (68) |
| Mediana, months (95% CI) | 9.3 (7.2, 16.6) | 7.4 (5.5, 9.9) | 2.0 (1.8, 2.3) |
| Hazard ratio (95% CI)b | 0.24c (0.17, 0.35) | 0.33d (0.24, 0.48) | N/A |
| p-value | <0.0001c | <0.0001d | N/A |
| Objective Response Rate (ORR) | |||
| Patients with measurable disease | 124 | 113 | 105 |
| Patients with CR or PR, n (%) | 39 (32) | 32 (28) | 1 (1.0) |
| 95% CI | 23, 40 | 20, 38 | 0, 5 |
| Duration of Response (DoR) | |||
| Mediana, months (95% CI) | 17.5 (8.8, NE) | 12.0 (8.1, NE) | NE (NE, NE) |
CI = confidence interval; CR = complete response; PR = partial response, NE = not estimable
a Based on Kaplan-Meier estimates.
b Stratified Cox regression model, confidence intervals for hazard ratio is based on the score test (D.Y. Lin 2016).
c Arm A (REVTORPYK plus fulvestrant and palbociclib) versus Arm C (fulvestrant).
d Arm B (REVTORPYK plus fulvestrant) versus Arm C (fulvestrant).
Figure 1. Kaplan-Meier Plot of Progression-Free Survival in Study 1 of VIKTORIA-1: REVTORPYK Plus Fulvestrant and Palbociclib versus Fulvestrant:
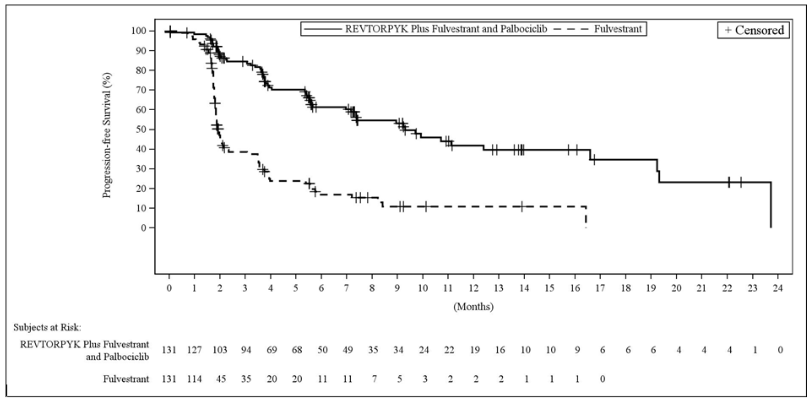
Figure 2. Kaplan-Meier Plot of Progression-Free Survival in Study 1 of VIKTORIA-1: REVTORPYK Plus Fulvestrant versus Fulvestrant:
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.