ROMVIMZA Capsule Ref.[115532] Active ingredients: Vimseltinib
Source: FDA, National Drug Code (US) Revision Year: 2025
12.1. Mechanism of Action
Vimseltinib is a kinase inhibitor that inhibits colony-stimulating factor 1 receptor (CSF1R). In vitro, vimseltinib inhibited CSF1R autophosphorylation, signaling induced by CSF1 ligand binding, and proliferation of cells expressing CSF1R.
12.2. Pharmacodynamics
Exposure-Response Relationship
Higher vimseltinib exposure is associated with an increased risk of all grades of edema, rash, increased AST, and increased ALT.
Vimseltinib exposure-response relationship for efficacy and time course of pharmacodynamic response have not been fully characterized.
Cardiac Electrophysiology
At the maximum recommended dose of ROMVIMZA, clinically significant QTc interval prolongation was not observed. However, the largest mean increase in QTc interval was 8.2 ms (upper confidence internal = 12.3 ms) after administration of vimseltinib 40 mg once daily for 5 days (3.3 times the maximum recommended weekly dose). The increase in QTc interval was concentration-dependent [see Clinical Pharmacology (12.3)].
12.3. Pharmacokinetics
Vimseltinib pharmacokinetic parameters were determined following a single oral dose of 30 mg or at steady state following multiple doses of 30 mg twice weekly and are provided as mean (CV%) unless otherwise specified.
Vimseltinib peak plasma concentration (Cmax) is 283 ng/mL (36%) or 747 ng/mL (39%) after a single dose or at steady state, respectively, and area under the time concentration curve (AUC0-inf) is 46,900 ng•h/mL (45%) after a single dose and AUC0-24hr is 13,400 ng•h/mL (45%) at steady state.
Vimseltinib pharmacokinetics are dose-proportional.
Absorption
Vimseltinib median time to Cmax (Tmax) is 1 hour (0.5 to 4 hours).
Effect of Food
No clinically significant differences in vimseltinib pharmacokinetics were observed following administration of a high-fat meal (800 to 1000 kcal, 50% fat), compared to fasted conditions.
Distribution
Vimseltinib volume of distribution (V/F) is 90 L (16%). Vimseltinib is 96.5% bound to human plasma proteins.
Elimination
Vimseltinib elimination half-life (t1/2) is approximately 6 days (32%) with a clearance (CL/F) of 0.5 L/h (23%).
Metabolism
Vimseltinib is primarily metabolized by oxidation, N-demethylation, and N-dealkylation; secondary biotransformation pathways included N-demethylation, dehydrogenation, and oxidation. CYP450 enzymes are not anticipated to play a major role in the metabolism of vimseltinib.
Excretion
Approximately 43% of the dose was recovered in feces (9.1% unchanged) and 38% in urine (5.1% unchanged) after a single radiolabeled dose.
Specific Populations
No clinically significant differences in the pharmacokinetics of vimseltinib were observed based on age (20 to 91 years), sex, race (Asian, Black or African American, White), body weight (43 to 150 kg), tumor (TGCT or other malignant solid tumors), and mild to moderate renal impairment (estimated glomerular filtration rate [eGFR] ≥30 mL/min calculated by CKD-EPI equation). The effect of severe renal impairment (eGFR <30 mL/min) or moderate to severe hepatic impairment (total bilirubin >1.5 x ULN with any AST) on vimseltinib pharmacokinetics is unknown.
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
P-glycoprotein (P-gp) inhibitors: Dabigatran (a P-gp substrate) AUC0-inf and Cmax are predicted to increase 2- to 3-fold with concomitant use with vimseltinib 30 mg twice weekly.
Dabigatran Cmax and AUC0-inf are predicted to increase up to 1.3-fold if administered 4 hours after administration of vimseltinib 30 mg twice weekly.
Other Drugs: No clinically significant differences in vimseltinib pharmacokinetics were observed when used concomitantly with itraconazole (a P-gp inhibitor) or rabeprazole (a proton pump inhibitor).
In Vitro Studies
CYP 450 enzymes: Vimseltinib is not a substrate of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A.
Vimseltinib is not an inhibitor of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4.
Vimseltinib is not an inducer of CYP1A2, CYP2B6, or CYP3A4.
Transporter systems: Vimseltinib is a P-gp substrate but is not a substrate of BCRP, BSEP, OATP1B1, OATP1B3, OAT1, OAT3, OCT2, MATE1, and MATE2-K.
Vimseltinib inhibits P-gp, BCRP, BSEP, OATP1B1, OATP1B3, OCT2, BSEP, MATE1, and MATE2-K. Vimseltinib does not inhibit OAT1 and OAT3. Vimseltinib may increase serum creatinine by decreasing renal tubular secretion of creatinine; this may occur due to inhibition of renal transporters OCT2 and MATE1 and may not affect renal function.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 6-month transgenic mouse carcinogenicity study at doses up to 12.5 mg/kg/day, vimseltinib was negative for carcinogenic findings.
Vimseltinib was not mutagenic in the bacterial reverse mutation assay (Ames). In an in vitro micronucleus assay, vimseltinib increased micronuclei after a 24-hour incubation in the absence of metabolic activation. In vivo, vimseltinib administered to rats at doses up to 200 mg/kg/day did not increase bone marrow micronucleated polychromatic erythrocytes, nor did vimseltinib increase liver DNA strand breaks.
In a fertility and early embryonic development study, male rats were administered 1, 2.5, or 5 mg/kg/day of vimseltinib starting 10 weeks before cohabitation, during cohabitation with untreated females, and at least 2 weeks post-cohabitation. Lower epididymal and testes weights were observed at 5 mg/kg/day (approximately 12 times the exposure at the recommended dose based on AUC). There were no treatment-related effects on mating, fertility, or sperm parameters at any dose tested. Female rats were administered 2.5, 5, or 10 mg/kg/day of vimseltinib 2 weeks prior to cohabitation with untreated males and during cohabitation until gestational day 7. Although there were no treatment-related effects on mating or estrous cycles, vimseltinib administered daily resulted in post-implantation loss at 10 mg/kg/day in female rats (approximately 20 times the exposure at the recommended dose based on AUC).
In a 26-week repeat-dose general toxicology study, recovery male rats that were administered 2.5 or 5 mg/kg/day had moderate to marked reductions in sperm and marked testicular atrophy (1 of 5 and 2 of 5 animals, respectively) corresponding to approximately 6 and 12 times the exposure at the recommended dose based on AUC, respectively. In a 39-week repeat-dose general toxicology study, minimal to moderate epididymal mineralization occurred in male dogs administered ≥4 mg/kg/day corresponding to exposures lower than the exposure at the recommended dose based on AUC.
13.2. Animal Toxicology and/or Pharmacology
In a 26-week repeat-dose general toxicology study in rats, chronic progressive nephropathy occurred at vimseltinib doses of ≥2.5 mg/kg/day (approximately 6 times the exposure at the recommended dose based on AUC). Degeneration of blood vessels (perivascular inflammation and necrosis of arteries and arteriole walls) occurred in multiple tissues at 5 mg/kg/day (approximately 12 times the exposure at the recommended dose based on AUC).
In a 39-week repeat-dose general toxicology study in dogs, skin depigmentation in the head and legs occurred at vimseltinib doses of ≥4 mg/kg/day corresponding to exposures lower than the exposure at the recommended dose based on AUC.
14. Clinical Studies
The efficacy of ROMVIMZA was evaluated in MOTION (NCT05059262), a phase 3, double-blind, multicenter, randomized (2:1), placebo-controlled study in patients with TGCT for whom surgical resection may cause worsening functional limitation or severe morbidity. Eligible patients had a confirmed diagnosis of TGCT with measurable disease per the Response Evaluation Criteria in Solid Tumors (RECIST v1.1) with at least one lesion having a minimum size of 2 cm. Patients were randomized to placebo or ROMVIMZA 30 mg twice weekly for 24 weeks. Randomization was stratified by tumor location (lower limb versus all other) and region (United States [US] versus non-US). At Week 25, patients who completed the double-blind, randomized part of the trial were eligible to advance to an ongoing, open-label extension study in which all patients received ROMVIMZA.
The major efficacy outcome measure was overall response rate (ORR) as assessed by blinded independent radiological review (IRR) per RECIST v1.1 at Week 25. Additional efficacy outcomes measured at Week 25 included ORR as assessed using tumor volume score (TVS), mean change from baseline in active range of motion of the affected joint at Week 25 measured by goniometry assessments, change from baseline in the Patient-Reported Outcomes Measurement Information System-Physical Function (PROMIS-PF) 15-item score (upper and lower extremity items), and response of at least a 30% improvement in the mean Brief Pain Inventory (BPI) Worst Pain numeric rating scale (NRS) score without a 30% or greater increase in narcotic analgesic use.
A total of 123 patients were randomized: 83 to ROMVIMZA and 40 to placebo during the double-blind period of the study. The median age was 44 years (range 20 to 78 years); 59% of patients were female; 65% were White, 4% were Asian, 3% were Black or African American, and 28% were not reported or unknown; 69% were not Hispanic or Latino, 3% were Hispanic or Latino, and 28% were not reported or unknown; 74% of patients had prior surgery; 69% of patients had diffuse TGCT; and 23% of patients were previously treated with systemic therapy. Disease locations were knee (67%), ankle (12%), hip (10%), other (5%), foot (3.3%), and wrist (2.4%).
A statistically significant improvement in ORR was demonstrated in patients randomized to ROMVIMZA compared with placebo. Efficacy results in MOTION are summarized in Table 6.
Table 6. Efficacy Results Assessed at Week 25 for MOTION:
| Efficacy Parameter | ROMVIMZA N=83 | Placebo N=40 |
|---|---|---|
| Overall Response Rate per RECIST v1.1 (95% CI) | 40% (29%, 51%) | 0% (0%, 9%) |
| Complete Response | 5% | 0% |
| Partial Response | 35% | 0% |
| p-value | <0.0001 | |
| Duration of Response1 | ||
| Median (Range in months)2 | NR (2.5+, 19.4+) | N/A |
| DOR ≥6 months | 28 (85%) | - |
| DOR ≥9 months | 19 (58%) | - |
| Active ROM3 | ||
| Patients with data at baseline and Week 25, n | 73 | 33 |
| Baseline mean (SD) | 63.0 (29.4) | 62.9 (32.2) |
| Mean change from baseline 4 (95% CI) | 18.4 (5.6, 31.2) | 3.8 (-10.5, 18.0) |
| Difference in LS means (95% CI) | 14.6% (4.0, 25.3) | |
| p-value | 0.0077 | |
| PROMIS-PF (15-Item score; ranges from 0-100)5 | ||
| Patients with data at baseline and Week 25, n | 63 | 30 |
| Baseline mean (SD) | 39.0 (6.1) | 38.5 (6.0) |
| Mean change from baseline 4 (95% CI) | 4.6 (2.7, 6.5) | 1.3 (-0.5, 3.0) |
| Difference in LS means (95% CI) | 3.3 (1.4, 5.2) | |
| p-value | 0.0007 | |
| BPI-30 Response6 | ||
| Patients with data at baseline and Week 25, n | 68 | 31 |
| Responders, n (%) | 40 (48.2%) | 9 (22.5%) |
| (95% CI) | (37.1%, 59.4%) | (10.8%, 38.5%) |
| Difference in responder rate (95% CI) 7 | 26.2% (9.5, 42.8) | |
| p-value | 0.0056 | |
NR=Not reached; N/A=Not applicable; AMA=American Medical Association; BPI=Brief Pain Inventory; CI=confidence interval; LS=least squares; MMRM=mixed model for repeated measures; n=number of patients in the category; N=sample size; PROMIS-PF=Patient-reported Outcomes Measurement Information System-Physical Function; ROM=range of motion; SD=Standard deviation.
1 DOR results are based on an additional 6 months of follow-up from the time of ORR analysis.
2 The median DOR was estimated using the Kaplan-Meier method. "+" indicates that the patient's response was ongoing at last assessment as of the data cutoff date.
3 Active ROM was normalized to the AMA reference standard.
4 Mean change from baseline was estimated from the MMRM for each corresponding endpoint. Baseline means presented include all patients and not only the ones with data at baseline and Week 25.
5 Data for PROMIS-PF is largely based on lower limb extremity assessment due to tumor location as described above. Higher scores of PROMIS-PF indicate better physical functioning.
6 BPI response in Worst Pain is defined as at least a 30% improvement in the mean BPI Worst Pain NRS score (0-10 NRS) without a 30% or greater increase in narcotic analgesic use at Week 25.
7 95% CI for the difference in response rates based on the stratified Mantel-Haenszel method.
ORR by TVS was 67% (95% CI: 56, 77) in patients randomized to ROMVIMZA and 0% in patients randomized to placebo; p-value <0.0001.
Individual patient results of change from baseline in active ROM at Week 25 (ROMVIMZA N=73; placebo N=33) are shown in Figure 1.
Figure 1. Change from Baseline in Active Range of Motion at Week 25 for MOTION:
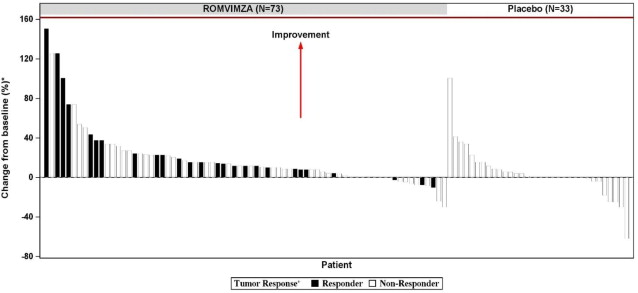
* Percent normal reference range for the affected joint.
+ Response by RECIST v1.1.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.