Source: Medicines & Healthcare Products Regulatory Agency (GB) Revision Year: 2021 Publisher: Roche Products Limited, 6 Falcon Way, Shire Park, Welwyn Garden City, AL7 1TW, United Kingdom
Casirivimab:
Pharmacotherapeutic group: Not yet assigned. ATC code: Not yet assigned.
Imdevimab:
Pharmacotherapeutic group: Not yet assigned. ATC code: Not yet assigned.
Casirivimab (IgG1κ) and imdevimab (IgG1λ) are two recombinant human monoclonal antibodies which are unmodified in the Fc regions. Casirivimab and imdevimab bind to non-overlapping epitopes of the spike protein receptor binding domain (RBD) of SARS-CoV-2 with dissociation constants KD = 45.8 pM and 46.7 pM, respectively. Casirivimab, imdevimab and casirivimab and imdevimab together blocked RBD binding to the human ACE2 receptor with IC50 values of 56.4 pM, 165 pM and 81.8 pM, respectively.
Casirivimab and imdevimab are intended to compensate/substitute for endogenous antibodies in those individuals who have yet to mount their own immune response.
There is a theoretical risk that antibody administration may attenuate the endogenous immune response to SARS-CoV-2 and make individuals more susceptible to re-infection.
Trial COV-2067 evaluated Ronapreve with doses up to 7 times the recommended dose (600 mg casirivimab and 600 mg imdevimab; 1,200 mg casirivimab and 1,200 g imdevimab; 4,000 mg casirivimab and 4,000 mg imdevimab) in ambulatory patients with COVID-19. A flat dose-response relationship for efficacy was identified for Ronapreve at all doses, based on viral load and clinical outcomes. Similar reductions in viral load (log10 copies/mL) were observed in subjects for the (600 mg casirivimab and 600 mg imdevimab) IV and (600 mg casirivimab and 600 mg imdevimab) subcutaneous doses.
COV-20145 is a Phase 2 randomised, double-blind, placebo-controlled, parallel group study to assess the dose response profile of single IV or single subcutaneous doses of Ronapreve in outpatients with SARS-CoV-2 infection. Treatment was initiated within 3 days of obtaining a positive SARS-CoV-2 infection test result in 803 adult patients not at high risk of severe disease (symptomatic with no risk factors/asymptomatic). Subjects were randomised into treatment arms and placebo arms including 116 subjects who were randomised to receive an IV dose of 1,200 mg of Ronapreve (600 mg of casirivimab and 600 mg of imdevimab).
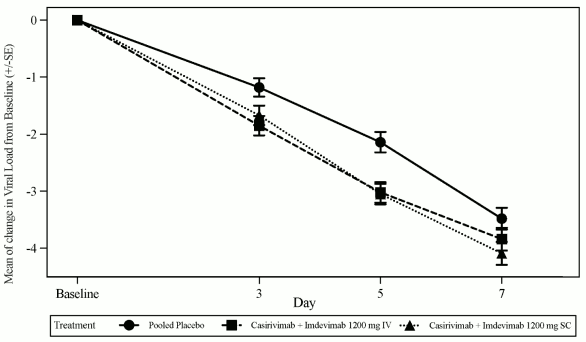
The pre-specified primary endpoint was the time weighted average (TWA) daily change from baseline in viral load (log10 copies/mL), as measured by RT-qPCR in nasopharyngeal swab samples, from Day 1 to Day 7 in subjects with a positive SARS-CoV-2 RT-qPCR result and seronegative at baseline i.e., the seronegative modified full analysis set (seronegative mFAS). Treatment with 1,200 mg IV Ronapreve resulted in a statistical significant reduction in the TWA from baseline to Day 7 in viral load compared to placebo (-0.56 log10 copies/mL, p<0.0007). The largest reductions in viral load relative to placebo occurred in patients with high viral load (>107 copies/mL) with a difference in TWA from Day 1 through Day 7 of -0.85 log10 copies/mL (p<0.0001). Figure 1 shows the mean change from baseline in SARS-CoV-2 viral load over time.
Figure 1. Mean Change in Viral Load (log10 copies/mL) at Each Visit from Baseline to Day 7 in Subjects Receiving 1,200 mg IV and 1,200 mg SC (Seronegative mFAS) Study COV20145:
The Phase 3 trial, COV-2067, is a randomised, double-blinded, placebo-controlled clinical trial evaluating Ronapreve (casirivimab and imdevimab) for the treatment of subjects with COVID-19 who are not hospitalised.
There were 4,567 adult subjects with at least one risk factor for severe COVID-19 were randomised to a single intravenous infusion of Ronapreve 1,200 mg (600 mg of casirivimab and 600 mg of imdevimab) (n=838), Ronapreve 2,400 mg (1,200 mg of casirivimab and 1,200 mg of imdevimab) (n=1 529), Ronapreve 8,000 mg (4,000 mg of casirivimab and 4,000 mg of imdevimab) (n=700), or placebo (n=1 500) groups. The two Ronapreve doses at the start of Phase 3 were 8,000 mg and 2,400 mg; however, based on Phase ½ efficacy analyses showing that the 8,000 mg and 2,400 mg doses were similar, the Phase 3 portion of the protocol was amended to compare 2,400 mg dose vs. placebo and 1,200 mg dose vs. placebo. Comparisons were between subjects randomised to the specific Ronapreve dose and subjects who were concurrently randomised to placebo.
The median age was 50 years (with 13% of subjects ages 65 years or older), 52% of the subjects were female, 84% were White, 5% were Black or African American; 36% identified as Hispanic or Latino.
The primary endpoint was the proportion of subjects with ≥ 1 COVID-19-related hospitalisation or allcause death through Day 29, in subjects with a positive SARS-CoV-2 RT-qPCR i.e., the modified full analysis set (mFAS), events (COVID-19-related hospitalisation or all-cause death through Day 29) occurred in 7 (1.0%) subjects treated with Ronapreve 1,200 mg compared to 24 (3%) subjects concurrently randomised to placebo, demonstrating a 70% relative risk reduction in COVID-19-related hospitalisation or all-cause death compared to placebo (p=0.0024).
In the 1,200 mg analysis, there was 1 death each in the Ronapreve and placebo arm (p=1.0); and in 2,400 mg analysis, there were 1 and 3 deaths, respectively, in the Ronapreve and placebo arms (p=0.3721). Overall, similar effects were observed for Ronapreve 1,200 mg (600 mg of casirivimab and 600 mg of imdevimab) and Ronapreve 2,400 mg (1,200 mg of casirivimab and 1,200 mg of imdevimab) doses, indicating the absence of a dose effect. Results were consistent across subgroups of patients defined by nasopharyngeal viral load >106 copies/mL at baseline.
The median time to symptom resolution, as recorded in a trial-specific daily symptom diary, was 10 days for Ronapreve treated subjects, as compared with 14 days for placebo-treated subjects (p=0.0001) for 1,200 mg vs. placebo; p<0.0001 for 2,400 mg vs. placebo). Treatment with Ronapreve resulted in a 4 days shorter median time to COVID-19 symptom resolution compared to placebo-treated subjects though cough, fatigue and/or headache may have persisted at reduced severity (i.e., mild or moderate per patient assessment).
Reduction in viral load was seen as early as the first post-baseline assessment, approximately two days after dosing. Treatment with Ronapreve resulted in a reduction in the LS mean viral load (log10 copies/mL) from baseline to Day 7 compared to placebo (-0.71 log10 copies/mL for Ronapreve 1,200 mg (600 mg dose of casirivimab and 600 mg of imdevimab) p<0.0001) and -0.86 log10 copies/mL for 2,400 mg; p<0.0001).
The data supporting prevention of COVID-19 are based on the efficacy analysis of data from the Phase 3 COV-2069 trial. This is a randomised, double-blind, placebo-controlled clinical trial studying Ronapreve (casirivimab and imdevimab) for prevention of COVID-19 in household contacts of individuals infected with SARS-CoV-2 (index case).
The trial enrolled adult subjects and 130 paediatric subjects aged 12 to 18 years who were asymptomatic and who lived in the same household with a SARS-CoV-2 infected patient. Subjects were randomised 1:1 to a single dose of Ronapreve 1,200 mg (600 mg of casirivimab and 600 mg of imdevimab) or placebo administered subcutaneously within 96 hours of collection of the index cases' positive SARS-CoV-2 diagnostic test sample. Subjects with a negative SARS-CoV-2 RT-qPCR test result, representing a mix of pre- and post-exposure prevention patients, joined Cohort A (2069-A). Subjects with a positive SARS-CoV-2 RT-qPCR test result, representing a cohort solely of postexposure prevention patients, joined Cohort B (2069-B). Baseline serology test results were used to further define analysis populations (seronegative subjects were considered not to have a prior infection whereas seropositive subjects were considered to have a prior infection).
Subjects with a negative SARS-CoV-2 RT-qPCR test result at baseline (n=2 067) were enrolled and randomised. The primary analysis population included subjects who were SARS-CoV-2 RT-qPCR negative and seronegative at baseline. Of the 1 505 subjects in the primary analysis population, 753 subjects were randomised to receive Ronapreve and 752 subjects were randomised to placebo. Following randomisation and dosing, subjects had SARS-CoV-2 RT-qPCR testing via a nasopharyngeal swab every 7 days as well as weekly interviews with the investigator for assessment of COVID-19 symptoms during the 28 day efficacy assessment period. No data were collected on the type or extent of exposure to the index case.
For the primary analysis population at baseline, the median age was 44 years (with 9% of subjects ages 65 years or older), 54% of the subjects were female, 86% were White, 9% were Black; 41% identified as Hispanic or Latino.
The primary efficacy endpoint in the primary analysis population was the proportion of subjects who developed symptomatic RT qPCR-confirmed COVID-19 through Day 29. In this population, there was a statistically significant 81% relative risk reduction in the development of COVID-19 with Ronapreve treatment versus placebo (see Table 5).
Table 5. Key Results from Phase 3 Trial for the Prevention of COVID-19 in Uninfected Individuals Study COV-2069, Cohort A:
| Ronapreve (single 1,200 mg dose) | Placebo | |
|---|---|---|
| Primary Analysis Population: Seronegative at Baseline | n=753 | n=752 |
| Risk of COVID-19 | ||
| Through Day 29 (primary endpoint) | ||
| Relative risk reduction (Odds ratio, p-Value) | 81% (0.17; p<0.0001) | |
| Number of individuals with events | 11 (1.5%) | 59 (7.8%) |
Asymptomatic subjects with a positive SARS-CoV-2 RT-qPCR test result at baseline (n=314) represent a post-exposure population. The primary analysis population included asymptomatic subjects who were SARS-CoV-2 RT-qPCR positive and seronegative at baseline. Of the 204 subjects in the primary analysis population, 100 subjects were randomised to receive Ronapreve and 104 subjects were randomised to placebo. Following randomisation and dosing, subjects had SARS-CoV-2 RT-qPCR testing via a nasopharyngeal swab every 7 days as well as weekly interviews with the investigator for assessment of COVID-19 symptoms during the 28 day efficacy assessment period. No data were collected on the type or extent of exposure to the index case.
For the primary analysis population at baseline, the median age was 40 years (with 11% of subjects ages 65 years or older), 55% of the subjects were female, 85% were White, 5% were Black; 35% identified as Hispanic or Latino.
The primary efficacy endpoint in the primary analysis population was the proportion of subjects who developed RT qPCR-confirmed COVID-19 through Day 29. There was a 31% relative risk reduction in the development of COVID-19 (see Table 6) with a more pronounced (76%) relative risk reduction in COVID-19 after Day 3, consistent with the disease progression being less modifiable within the first days of treatment. Similar results were obtained in the sensitivity analysis that included all RT-qPCR positive subjects at baseline, regardless of baseline serological status, where there was a 35% relative risk reduction in PT-qPCR-confirmed COVID-19 with Ronapreve treatment compared to placebo. Ronapreve also reduced the total number of symptomatic weeks, number of high viral load weeks, and number of subjects who require hospitalisation or emergency room visits.
Table 6. Key Results in Asymptomatic Infected Individuals Study COV-2069, Cohort B:
| Ronapreve (single 1,200 mg dose) | Placebo | |
|---|---|---|
| Primary Analysis Population: Seronegative at Baseline | n=100 | n=104 |
| Risk of COVID-19 | ||
| Overall relative risk reduction through Day 29 (primary endpoint) | ||
| Relative risk reduction (Odds ratio, p-Value) | 31% (0.54; p=0.0380) | |
| Number of individuals with events | 29 (29%) | 44 (42.3%) |
The data supporting the use for the repeat dose prevention of COVID-19 are based on the exploratory efficacy analysis of data from 969 subjects from the Phase 1 HV-2093. HV-2093 is a randomised, double-blind, placebo-controlled Phase 1 clinical trial assessing the safety, tolerability, pharmacokinetics, and immunogenicity of repeat subcutaneous doses (up to 6 monthly doses) of Ronapreve (casirivimab with imdevimab) in adult subjects who are SARS-CoV-2 negative at baseline. Subjects were randomised in a 3:1 manner to receive subcutaneous injections every 4 weeks for 24 weeks of 1,200 mg of Ronapreve (600 mg casirivimab and 600 mg imdevimab) (n=729) or placebo (n=240).
At baseline, the median age was 48 years (with 13% of subjects ages 65 years or older), 55% of the subjects were male, 87% were White, 10% were Black; 23% identified as Hispanic or Latino. The baseline demographics and disease characteristics were well balanced across the Ronapreve and placebo treatment groups.
The primary purpose of the study was PK (see section 5.2). An efficacy endpoint was the incidence of clinically diagnosed COVID-19. During the six-month treatment period, there was a 92% relative risk reduction in COVID-19, with Ronapreve treatment versus placebo: 3/729 (0.4%) versus 12/240 (5.0%), respectively; odds ratio (OR) 0.08 (95% CI: 0.01, 0.30); nominal p<0.0001. Of the subjects who developed COVID-19, 9/12 placebo recipients had a positive SARS-CoV-2 RT-PCR result or seroconverted whereas 0/3 subjects in the Ronapreve group were RT-PCR positive or seroconverted by the end of the treatment period.
In all subjects who received Ronapreve by intravenous infusion or subcutaneous injection, the incidence of anti-casirivimab and anti-imdevimab antibodies were 0.8% and 1.7%, respectively. For subjects who received placebo, the incidence of anti-casirivimab and anti-imdevimab antibodies were 1.9% and 4.5%, respectively.
In 707 subjects treated with Ronapreve 1,200 mg (600 mg of casirivimab and 600 mg of imdevimab) subcutaneously every 4 weeks, the incidence of treatment-emergent anti-casirivimab and antiimdevimab antibodies was 0.1% and 2.0%, respectively. Among 232 repeat dose placebo subjects, the incidence of treatment emergent anti-casirivimab and anti-imdevimab antibodies were 0% and 2.6%, respectively. The antibody titers in both REGEN-COV and placebo repeat dose subjects were low, with no evidence of altered pharmacokinetic profiles of casirivimab or imdevimab.
All studies enrolled adult patients; study 2069 also enrolled subjects aged 12-18 years; subjects aged <12 years have not been studied.
All subjects were enrolled in the community; none was enrolled when in hospital; none was receiving supplemental oxygen to treat acute covid infection.
The lightest body weight of all subjects was 35.5 kg.
Both casirivimab and imdevimab exhibited linear and dose-proportional pharmacokinetics (PK) between 300 mg Ronapreve (150 mg casirivimab and 150 mg imdevimab) to 8,000 mg Ronapreve (4,000 mg casirivimab and 4 000 mg imdevimab) following IV administration of single dose. A summary of PK parameters after a single (600 mg casirivimab and 600 mg imdevimab) IV dose, calculated using a population PK model for each antibody based on data from 3,687 subjects (casirivimab) or 3,716 subjects (imdevimab), is provided in Table 7.
Table 7. Summary of PK Parameters (for casirivimab and imdevimab) After a Single 1,200 mg IV Dose of Ronapreve:
| PK Parameter1 | casirivimab | imdevimab |
|---|---|---|
| AUC0-28 (mg·day/L)2 | 1754.9 (380.50) | 1600.8 (320.88) |
| AUCinf (mg·day/L)3 | 3563.6 (1239.61) | 2890.5 (876.31) |
| Cmax (mg/L)4 | 182.7 (81.45) | 181.7 (77.78) |
| C28 (mg/L)5 | 37.9 (10.33) | 31.0 (8.24) |
| Half-life (day) | 31.2 (10.59) | 27.3 (7.73) |
1 Mean (SD), where SD is standard deviation of the arithmetic mean; 2AUC0-28 = Area under the concentration time curve from time 0 to 28 days after dosing; 3AUCinf = Area under the concentration time curve from time 0 to infinite time; 4Cmax = Maximum concentration in serum and represents concentration at the end of infusion; 5C28 = Concentration 28 days after dosing, i.e., on day 29
A summary of PK parameters after a single Ronapreve 1,200 mg (600 mg casirivimab and 600 mg imdevimab) subcutaneous dose based on the population PK model for each antibody is shown in Table 8.
Table 8. Summary of PK Parameters for casirivimab and imdevimab after a Single 1,200 mg Subcutaneous Dose of Ronapreve:
| PK Parameter1 | casirivimab | imdevimab |
|---|---|---|
| AUC0-28 (mg·day/L)2 | 1121.7 (243.12) | 1016.9 (203.92) |
| AUCinf (mg·day/L)3 | 2559.5 (890.35) | 2073.3 (628.60) |
| Cmax (mg/L)4 | 52.2 (12.15) | 49.2 (11.01) |
| tmax (day)5,6^ | 6.7 [3.4, 13.6] | 6.6 [3.4, 13.6] |
| C28 (mg/L)7 | 30.5 (7.55) | 25.9 (6.07) |
1 Mean (SD), where SD is standard deviation of the arithmetic mean; 2AUC0-28 = Area under the concentration time curve from time 0 to 28 days after dosing; 3AUCinf = Area under the concentration time curve from time 0 to infinite time; 4Cmax = Maximum concentration in serum; 5tmax = Time to reach Cmax; 6Median [minimum, maximum]; 7C28 = Concentration 28 days after dosing, i.e., on day 29
A summary of PK parameters after a single 1,200 mg intravenous loading dose of Ronapreve (600 mg casirivimab and 600 mg imdevimab) followed by multiple 600 mg Ronapreve intravenous Q4W doses (300 mg casirivimab and 300 mg imdevimab) based on the population PK model for each antibody is shown in Table 9.
Table 9. Summary of PK Parameters for casirivimab and imdevimab after a Single 1,200 mg IV Loading Dose and 600 mg IV Q4W Maintenance Doses of Ronapreve:
| PK Parameter1 | casirivimab | imdevimab |
|---|---|---|
| AUCtau,ss(mg∙day/L)2 | 1767.5 (605.79) | 1436.8 (432.87) |
| Cmax,ss (mg/L)3 | 133.8 (46.51) | 122.4 (41.67) |
| Ctrough,ss (mg/L)4 | 42.6 (19.72) | 31.7 (13.56) |
| C28 (mg/L)5 | 37.9 (10.32) | 31.0 (8.24) |
| AR6 | 1.0 (0.241) | 0.893 (0.174) |
1 Mean (SD), where SD is standard deviation of the arithmetic mean; 2AUCtau,ss = Area under the concentration time curve during a dosing interval at steady-state; 3Cmax,ss = Maximum concentration at steady-state; 4Ctrough,ss = Trough concentration at steady-state; 5C28 = Concentration 28 days after the first dose; 6The accumulation ratio (AR) is calculated as AUCτ,ss / AUCτ,FD (FD = first dose); Q4W = Every 4 weeks
A summary of PK parameters after a single subcutaneous 1,200 mg loading dose of Ronapreve (600 mg casirivimab and 600 mg imdevimab) followed by multiple subcutaneous Q4W doses of 600 mg Ronapreve (300 mg casirivimab and 300 mg imdevimab) based on the population PK model for each antibody is shown in Table 10.
Table 10. Summary of PK Parameters for casirivimab and imdevimab after a Single 1,200 mg Subcutaneous Loading Dose and 600 mg Subcutaneous Q4W Maintenance Doses of Ronapreve:
| PK Parameter1 | casirivimab | imdevimab |
|---|---|---|
| AUCtau,ss (mg∙day/L)2 | 1268.9 (434.68) | 1030.1 (310.30) |
| Cmax,ss (mg/L)3 | 56.0 (16.81) | 47.0 (12.43) |
| Ctrough,ss (mg/L)4 | 34.0 (14.56) | 26.1 (10.17) |
| C28 (mg/L)5 | 30.5 (7.55) | 25.9 (6.07) |
| AR6 | 1.13 (0.288) | 1.01 (0.213) |
1 Mean (SD), where SD is standard deviation of the arithmetic mean; 2AUCtau,ss = Area under the concentration time curve during a dosing interval at steady-state; 3Cmax,ss = Maximum concentration at steady-state; 4Ctrough,ss = Trough concentration at steady-state; 5C28 = Concentration 28 days after the first dose; 6The accumulation ratio (AR) is calculated as AUCτ,ss / AUCτ,FD (FD = first dose); Q4W = Every 4 weeks
For the repeat dose prevention of IV and subcutaneous regimens, population pharmacokinetic simulations predict that median predicted casirivimab and imdevimab Ctrough,ss in serum are similar to observed mean day 29 concentrations in serum for a single subcutaneous dose of Ronapreve 1,200 mg (600 mg of casirivimab and 600 mg of imdevimab).
Based on population pharmacokinetic modeling, mean (standard deviation) Cmax and C28 estimates for casirivimab and imdevimab following single IV or single subcutaneous dose 1,200 mg (600 mg each monoclonal antibody) are listed in Table 7 and Table 8, respectively. Median (range) time to reach maximum serum concentration of casirivimab and imdevimab (Tmax) estimates following a single subcutaneous dose of Ronapreve 1,200 mg (600 mg each monoclonal antibody) are 6.8 (3.4-13.6) days and 6.6 (3.4-13.6) days for casirivimab and imdevimab, respectively (Table 8). Following casirivimab and imdevimab administered as a single dose of Ronapreve 1,200 mg subcutaneous (600 mg each monoclonal antibody), casirivimab and imdevimab had a population PK estimated bioavailability of 71.8% and 71.7%, respectively.
The total volume of distribution estimated via population pharmacokinetic analysis is 7.161 L and 7.425 L for casirivimab and imdevimab, respectively.
Specific metabolism studies were not conducted because casirivimab and imdevimab are proteins. As human monoclonal IgG1 antibodies, casirivimab and imdevimab are expected to be degraded into small peptides and amino acids via catabolic pathways in the same manner as endogenous IgG.
Based on population PK analysis, the terminal elimination half-life and clearance of casirivimab and imdevimab are listed in Table 11.
Table 11. Summary of Terminal Elimination Half-Life and Clearance Values of casirivimab and imdevimab Following Single IV Doses – Population PK Estimates:
| casirivimab | imdevimab | |||
|---|---|---|---|---|
| Parameter | Mean | 5th, 95th percentile | Mean | 5th, 95th percentile |
| Half-life (day) | 29.8 | (16.4, 43.1) | 26.2 | (16.9, 35.6) |
| CL (L/day) | 0.182 (2.21% RSE) | (0.11, 0.3) | 0.221 (1.87% RSE) | (0.15, 0.35) |
Casirivimab and imdevimab are monoclonal antibodies and are therefore not likely to undergo renal excretion.
Adolescent subjects (≥12 years of age and ≥40 kg) were enrolled in studies (COV-2067, COV-2069) however no PK data were available in these subjects. Since adolescents' body weight range is generally within the range of body weight in adult subjects and generally body weight is the main covariate that affects exposure in this age range, exposures of casirivimab and imdevimab in adolescent subjects (≥40kg) are expected to be similar to those in adults. The pharmacokinetics of casirivimab and imdevimab in pediatric patients (<12 years) have not been established.
The minimum body weight of subjects in clinical studies was 35.5 kg. There is no experience of use in subjects at lower body weight where AUC and Cmax are predicted to be at least 30% higher.
In the population PK analysis, age (18 years to 96 years) was not identified as a significant covariate on PK of either casirivimab and imdevimab.
Compared to patients <65 years of age, exposures of casirivimab and imdevimab were similar in patients who were aged >65 years or ≥75 years after either IV or subcutaneous administration.
Casirivimab and imdevimab are monoclonal antibodies that are not expected to undergo significant renal elimination due to their molecular weight (>69 kDa). Based on population PK analysis, trough concentrations of casirivimab and imdevimab in serum at steady state were comparable between patients with mild or moderate renal impairment, or patients with CrCl <15 mL/min including those on dialysis, and patients with normal renal function. Limited data are available in patients with severe renal impairment (n=3).
Casirivimab and imdevimab are not expected to undergo significant hepatic elimination. The effect of hepatic impairment on the exposure of casirivimab and imdevimab was evaluated by population PK analysis in patients with mild hepatic impairment (n=586 for casirivimab and n=599 for imdevimab) (total bilirubin [TB] greater than 1.0 to 1.5 times the upper limit of normal [ULN] and any aspartate aminotransferase [AST]); no clinically important differences in the exposure of casirivimab and imdevimab were found between patients with mild hepatic impairment and patients with normal hepatic function. Limited data (n=11) are available in patients with moderate hepatic impairment. The pharmacokinetics in patients with severe hepatic impairment has not been studied
A population PK analysis suggests that the following factors have no clinically significant effect on the exposure of casirivimab and imdevimab: age, gender, body weight, race, albumin level, renal impairment, and mild hepatic impairment.
Compared to a reference 81 kg subject, exposures (AUCday28, Cmax and Cday28) are predicted to be 20-30% higher in subjects at the 5th percentile of body weight (55.4 kg) and 20-25% lower in subjects at the 95th percentile of body weight (123 kg) for both casirivimab and imdevimab.
Compared to a reference 81 kg subject, the subject with the combination of covariates leading to the highest population predicted casirivimab-imdevimab CL (White, male, albumin 29 g/L, 151.8 kg) is predicted to have AUCday28, Cmax, and Cday28 ratios of 0.48, 0.56, and 0.31 respectively for casirivimab, and 0.47, 0.56, and 0.28 respectively for imdevimab.
Carcinogenicity, genotoxicity, and reproductive toxicology studies have not been conducted with casirivimab and imdevimab.
In a toxicology study in cynomolgus monkeys, casirivimab and imdevimab had no adverse effects when administered intravenously or subcutaneously. Non-adverse liver findings (minor transient increases in AST and ALT) were observed.
In tissue cross-reactivity studies with casirivimab and imdevimab using human and monkey adult tissues and human foetal tissues, no binding was detected.
For the single-dose treatment or acute prevention indications, when the estimated AUCcum at the NOAEL in the 4-week toxicology study is compared to the predicted AUCinf in human subjects, the exposure multiples are approximately 37.5 and 52.3 for Ronapreve 1,200 mg (600 mg of casirivimab and 600 mg of imdevimab) IV and 1,200 mg (600 mg of casirivimab and 600 mg of imdevimab) subcutaneous, respectively.
For the repeat-dose chronic prevention indications, when the estimated 4-week AUC following the last dose at the NOAEL in the 4-week toxicology study (AUCtau,ss) is compared to the predicted AUCtau,ss in human subjects, the exposure multiples are approximately 35.3 and 49.1 for Ronapreve 1,200 mg (600 mg of casirivimab and 600 mg of imdevimab) IV and 1,200 mg (600 mg of casirivimab and 600 mg of imdevimab) subcutaneous loading doses, followed by Ronapreve 600 mg (300 mg of casirivimab and 300 mg of imdevimab) Q4W dosing regimens, respectively.
There is a potential risk of treatment failure due to the development of viral variants that are resistant to casirivimab and imdevimab. Prescribing healthcare providers should consider the prevalence of SARS-CoV-2 variants in their area, where data are available, when considering treatment options.
Based on in vitro testing, casirivimab and imdevimab in combination are expected to retain neutralization potency against the following variants of concern/interest: B.1.1.7 (UK origin/Alpha, B.1.351 (South Africa origin/Beta, P.1, B.1.427/B.1.429 (California origin/Epsilon), B.1.526 (New York origin/Iota), B.1.617.1/B.1.617.3 (India origin/Kappa) and B.1.617.2 (India origin/Delta); however, it is not known how in vitro neutralisation data correlate with clinical outcomes.
There is a potential risk of treatment failure due to the development of viral variants that are resistant to casirivimab and imdevimab. Prescribing healthcare providers should consider the prevalence of SARS-CoV-2 variants in their area, where data are available, when considering treatment options.
Based on in vitro testing, casirivimab and imdevimab in combination are expected to retain neutralization potency against the following variants of concern/interest: B.1.1.7 (UK origin/Alpha), B.1.351 (South Africa origin/Beta), P.1 (Brazil origin/Gamma), B.1.427/B.1.429 (California origin/Epsilon), B.1.526 (New York origin/Iota), B.1.617.1/B.1.617.3 (India origin/Kappa) and B.1.617.2 (India origin/Delta), C.37 (Peru origin/Lambda) and AY.1/AY.2 (India origin/Delta), however, it is not known how in vitro neutralization data correlate with clinical outcomes.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.