RUBRACA Film-coated tablet Ref.[7613] Active ingredients: Rucaparib
Source: European Medicines Agency (EU) Revision Year: 2025 Publisher: pharmaand GmbH, Taborstrasse 1, 1020 Vienna, Austria
Pharmacodynamic properties
Pharmacotherapeutic group: Other antineoplastic agents
ATC code: L01XK03
Mechanism of action and pharmacodynamics effects
Rucaparib is an inhibitor of poly(ADP-ribose) polymerase (PARP) enzymes, including PARP-1, PARP-2, and PARP-3, which play a role in DNA repair. In vitro studies have shown that rucaparib-induced cytotoxicity involves inhibition of PARP enzymatic activity and the trapping of PARP-DNA complexes resulting in increased DNA damage, apoptosis, and cell death.
Rucaparib has been shown to have in vitro and in vivo anti-tumour activity in BRCA mutant cell lines through a mechanism known as synthetic lethality, whereby the loss of two DNA repair pathways is required for cell death. Increased rucaparib-induced cytotoxicity and anti-tumour activity was observed in tumour cell lines with deficiencies in BRCA1/2 and other DNA repair genes. Rucaparib has been shown to decrease tumour growth in mouse xenograft models of human cancer with or without deficiencies in BRCA.
Clinical efficacy
First-Line maintenance treatment of advanced ovarian cancer
The efficacy of rucaparib was evaluated in ATHENA, a Phase 3, double-blind, multicentre trial in which 538 patients with advanced ovarian (EOC), fallopian tube (FTC), or primary peritoneal cancer (PPC) who were in response to first-line platinum-based chemotherapy and surgery were enrolled. Response was defined as no evidence of disease progression radiologically or through rising CA-125 (per Gynecological Cancer Intergroup (GCIG) guidelines) at any time during first-line treatment; and either no evidence of measurable disease by RECIST v1.1, if complete resection after surgery, or a response (complete or partial) if measurable disease was present after surgery and prior to chemotherapy, or a GCIG CA-125 response if non-measurable disease was present in the same situation.
All patients had received between 4 to 8 cycles of platinum-doublet treatment (including ≥4 cycles of platinum/taxane combination). Bevacizumab treatment was allowed during first-line chemotherapy, but not during the maintenance rucaparib treatment. All patients were randomised within 8 weeks of the first day of the last cycle of chemotherapy.
Patients were randomised (4:1) to receive rucaparib tablets 600 mg orally twice daily (n=427) or placebo (n=111). Treatment was continued until disease progression or unacceptable toxicity or for up to 2 years. Randomisation was stratified by disease status post-chemotherapy (residual disease vs. no residual disease), timing of surgery (primary surgery vs. interval debulking), and biomarker status. Biomarker status was determined using the homologous recombination deficiency (HRD) test where biomarker-positive was a tumour with HRD defined by the presence of a deleterious tumour BRCA (tBRCA) mutation or tBCRA wild type (tBRCAwt)/high genomic loss of heterozygosity (LOHhigh), and biomarker-negative was a tumour without HRD, defined by tBRCAwt/low genomic LOH (LOHlow).
The major efficacy outcome was investigator-assessed progression-free survival (invPFS) evaluated according to Response Evaluation Criteria in Solid Tumours (RECIST), version 1.1. Key Secondary efficacy endpoints included overall survival (OS) and objective response rate (ORR) according to RECIST version 1.1. invPFS, OS and ORR testing were performed hierarchically: first in the HRD Group, then in the ITT population. Time from randomisation to second progression or death (PFS2), was an additional outcome measure.
The median age of patients treated with rucaparib was 61 years (range: 30 to 83) and 62 years (range: 31 to 80) among patients on placebo. The Eastern Cooperative Oncology Group (ECOG) performance status was 0 in 69% of patients receiving rucaparib and 68% of patients receiving placebo. Of the 538 patients randomised to rucaparib or placebo, 75% had FIGO Stage III disease and 25% had Stage IV disease, and 16% were in complete response to their most recent platinum-based regimen. Of the 538 patients randomised to rucaparib or placebo 78% had EOC, 13% had FTC and 9% had PPC, most patients (>90%) had tumours with serous histology. In the ITT population, patients received a median of 6 cycles of platinum doublet chemotherapy and 17.8% of patients had received bevacizumab during first-line chemotherapy. Primary debulking surgery had been performed in 48.1% of patients, and 51.9% of patients had undergone neo-adjuvant chemotherapy followed by interval debulking surgery.
Overall, 43% had HRD (21% had a deleterious tBRCA mutation and 22% had tBRCAwt/LOHhigh), 44% were HRD negative (tBRCAwt/LOHlow), and 12% had an unknown HRD status.
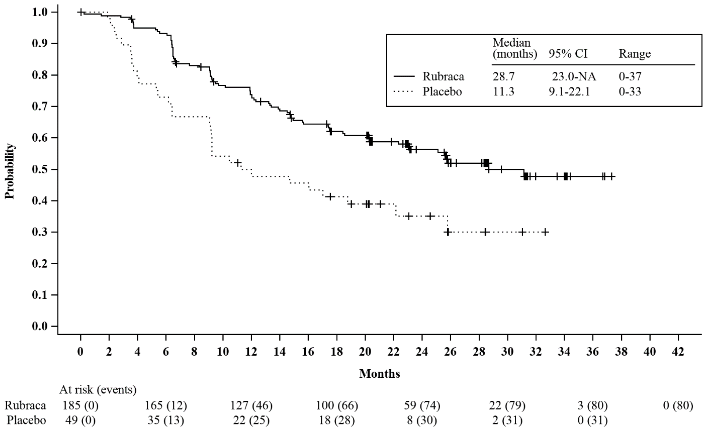
ATHENA demonstrated a statistically significant improvement in invPFS for patients randomised to rucaparib as compared with placebo in the HRD Group and the ITT Population. Results for invPFS with and without censoring for new anti-cancer treatment and missed visits were consistent. Efficacy results are presented in Table 4 and Figures 1 and Figure 2.
Table 4. Efficacy Results – ATHENA (Investigator Assessment):
| HRD Groupa | ITT Populationb | |||
|---|---|---|---|---|
| Rubraca (n=185) | Placebo (n=49) | Rubraca (n=427) | Placebo (n=111) | |
| PFSc events, n (%) | 80 (43.2) | 31 (63.3) | 230 (53.9) | 78 (70.3) |
| PFS median in months (95% CI) | 28.7 (23.0, NR) | 11.3 (9.1,22.1) | 20.2 (15.2,24.7) | 9.2 (8.3,12.2) |
| Hazard ratio (95% CI) | 0.47 (0.31, 0.72) | 0.52 (0.40, 0.68) | ||
| P-valued | 0.0005 | <0.0001 | ||
| OSe events, n (%) | 46 (24.9) | 12 (24.5) | 144 (33.7) | 42 (37.8) |
| OS median in months | NR | NR | NR | 46.2 |
| Hazard ratio (95% CI) | 0.84 (0.44, 1.58) | 0.83 (0.58, 1.17) | ||
| P-valued | 0.5811 | 0.2804 | ||
a Includes all patients with a deleterious tBRCA mutation (N=115) or tBRCAwt/LOHhigh (N=119).
b All randomised patients.
c The median follow up time was 26 months for both the rucaparib and placebo arms.
d P-value based on the stratified logrank test.
e At the time of the second interim analysis, the OS data were not mature (35% of patients had died); the median follow up time was 37 months for both the rucaparib and placebo arms.
NR: Not reached.
Figure 1. Kaplan-Meier Curves of Progression-Free Survival in ATHENA as Assessed by Investigator: ITT Population:
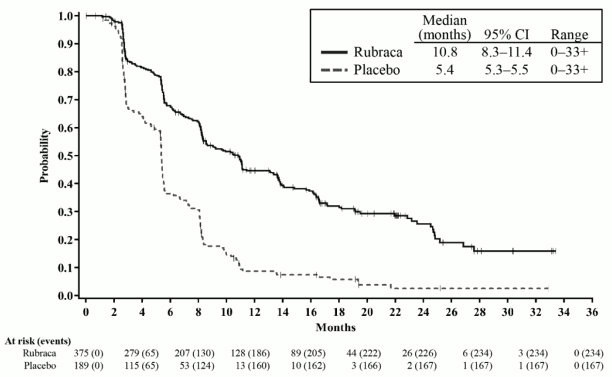
Figure 2. Kaplan-Meier Curves of Progression-Free Survival in ATHENA as Assessed by Investigator: HRD Population:
Subgroup analysis (PFS investigator assessment)
Within the HRD Population, a hazard ratio of 0.40 (95% CI [0.21, 0.75]) was observed in the subgroup of patients with a tBRCA mutation (n=115). In the subgroup of non-tBRCA LOHhigh (n=119), a hazard ratio of 0.58 (95% CI [0.33, 1.01]). In the HRD-negative subgroup (n=238), a hazard ratio of 0.65 (95% CI [0.45, 0.95]) was observed.
Maintenance treatment of recurrent ovarian cancer
The efficacy of rucaparib was investigated in ARIEL3, a double-blind, multicentre clinical trial in which 564 patients with recurrent EOC, FTC or PPC who were in response to platinum-based chemotherapy were randomised (2:1) to receive Rubraca tablets 600 mg orally twice daily (n=375) or placebo (n=189). Treatment was continued until disease progression or unacceptable toxicity. All patients had achieved a response (complete or partial) to their most recent platinum-based chemotherapy and their cancer antigen 125 (CA-125) was below the upper limit of normal (ULN). Patients were randomised within 8 weeks of completion of platinum chemotherapy and no intervening maintenance treatment was permitted. Patients could not have received prior rucaparib or other PARP inhibitor therapy. Randomisation was stratified by best response to last platinum therapy (complete or partial), time to progression following the penultimate platinum therapy (6 to ≤12 months and >12 months), and tumour biomarker status (tBRCA, non-BRCA homologous recombination deficiency [nbHRD] and biomarker negative).
The primary efficacy outcome was invPFS evaluated according to RECIST, version 1.1 (v1.1). PFS assessed by blinded independent radiology review (IRR) was a key secondary efficacy outcome. Secondary efficacy endpoints included overall survival (OS).
The mean age was 61 years (range: 36 to 85 years); most of the patients were White (80%); and all had an ECOG performance status of 0 or 1. The primary tumour in most patients was ovarian (84%); most patients (95%) had serous histology and 4% of patients reported endometrioid histology. All patients had received at least two prior platinum-based chemotherapies (range: 2 to 6) and 28% of patients had received at least three prior platinum-based chemotherapies. A total of 32% of patients were in complete response (CR) to their most recent therapy. The progression-free interval to penultimate platinum therapy was 6-12 months in 39% of patients and >12 months in 61%. Prior bevacizumab therapy was reported for 22% of patients who received rucaparib and 23% of patients who received placebo. Demographics, baseline disease characteristics, and prior treatment history were generally well balanced between the rucaparib and placebo arms.
None of the patients had received prior treatment with a PARP inhibitor. As such, efficacy of Rubraca in patients who have received prior treatment with a PARP inhibitor in the maintenance setting, has not been investigated and cannot be extrapolated from the available data.
Tumour tissue samples for all of the patients (N=564) were tested centrally to determine HRD positive status (as defined by the presence of a deleterious tumour BRCA [tBRCA] mutation or high genomic loss of heterozygosity). Blood samples for 94% (186/196) of the tBRCA patients were evaluated using a central blood germline BRCA (gBRCA) test. Based on these results, 70% (130/186) of the tBRCA patients had a gBRCA mutation and 30% (56/186) had a somatic BRCA mutation.
The ARIEL3 study met its primary endpoint and demonstrated a statistically significant improvement in invPFS for patients randomised to rucaparib as compared with placebo in the ITT population and in the HRD and tBRCA groups. IRR-assessment for the ITT population supported the primary endpoint. PFS results are summarised in Table 5 and Figure 3.
Table 5. ARIEL3 Efficacy Results (Summary of Primary Objective Outcome: PFS):
| Parameter | Investigator Assessment | IRR | ||
|---|---|---|---|---|
| Rucaparib | Placebo | Rucaparib | Placebo | |
| ITT populationa | ||||
| Patients, n | 375 | 189 | 375 | 189 |
| PFS events, n (%) | 234 (62%) | 167 (88%) | 165 (44%) | 133 (70%) |
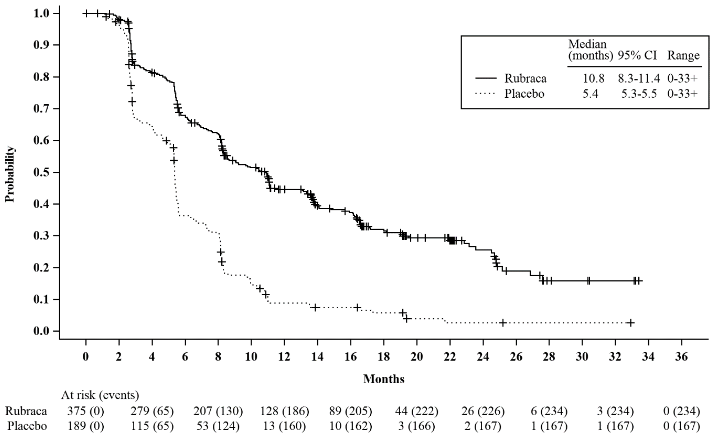
| PFS, median in months (95% CI) | 10.8 (8.3, 11.4) | 5.4 (5.3-5.5) | 13.7 (11.0, 19.1) | 5.4 (5.1, 5.5) |
| HR (95% CI) | 0.36 (0.30, 0.45) | 0.35 (0.28, 0.45) | ||
| p-valueb | <0.0001 | <0.0001 | ||
| HRD Groupc | ||||
| Patients, n | 236 | 118 | 236 | 118 |
| PFS events, n (%) | 134 (57%) | 101 (86%) | 90 (38%) | 74 (63%) |
| PFS, median in months (95% CI) | 13.6 (10.9, 16.2) | 5.4 (5.1, 5.6) | 22.9 (16.2, NA) | 5.5 (5.1, 7.4) |
| HR (95% CI) | 0.32 (0.24, 0.42) | 0.34 (0.24, 0.47) | ||
| p-valueb | <0.0001 | <0.0001 | ||
| tBRCA Groupd | ||||
| Patients, n | 130 | 66 | 130 | 66 |
| PFS events, n (%) | 67 (52%) | 56 (85%) | 42 (32%) | 42 (64%) |
| PFS, median in months (95% CI) | 16.6 (13.4, 22.9) | 5.4 (3.4, 6.7) | 26.8 (19.2, NA) | 5.4 (4.9, 8.1) |
| HR (95% CI) | 0.23 (0.16, 0.34) | 0.20 (0.13, 0.32) | ||
| p-valueb | <0.0001 | <0.0001 | ||
| nonBRCA LOH+ Group | ||||
| Patients, n | 106 | 52 | 106 | 52 |
| PFS events, n (%) | 67 (63%) | 45 (87%) | 48 (45%) | 32 (62%) |
| PFS, median in months (95% CI) | 9.7 (7.9, 13.1) | 5.4 (4.1, 5.7) | 11.1 (8.2, NA) | 5.6 (2.9, 8.2) |
| HR (95% CI) | 0.44 (0.29, 0.66) | 0.554 (0.35, 0.89) | ||
| p-valueb | <0.0001 | 0.0135 | ||
| nonBRCA LOH- Group | ||||
| Patients, n | 107 | 54 | 107 | 54 |
| PFS events, n (%) | 81 (73%) | 50 (93%) | 63 (59%) | 46 (85%) |
| PFS, median in months (95% CI) | 6.7 (5.4, 9.1) | 5.4 (5.3, 7.4) | 8.2 (5.6, 10.1) | 5.3 (2.8, 5.5) |
| HR (95% CI) | 0.58 (0.40, 0.85) | 0.47 (0.31, 0.71) | ||
| p-valueb | 0.0049 | 0.0003 | ||
a All randomised patients.
b Two-sided p-value
c HRD includes all patients with a deleterious germline or somatic BRCA mutation or non-tBRCA with high genomic loss of heterozygosity, as determined by the clinical trial assay (CTA).
d tBRCA includes all patients with a deleterious germline or somatic BRCA mutation, as determined by the CTA.
HR: Hazard ratio. A value <1 favours rucaparib.
NA: Not Achieved
CI: Confidence interval
Figure 3. Kaplan-Meier Curves of Progression-Free Survival in ARIEL3 as Assessed by Investigator: ITT population:
At the final OS analysis (70% maturity) the Hazard Ratio (HR) was 1.00 (95% CI: 0.81, 1.22; median 36 months for rucaparib vs 43.2 months for placebo) for the ITT population. For the HRD and tBRCA subgroups the reported HRs were 1.01 (95% CI: 0.77, 1.32; median 40.5 months for rucaparib vs 47.8 months for placebo) and 0.83 (95% CI: 0.58, 1.19; median 45.9 months for rucaparib vs 47.8 months for placebo), respectively. In an exploratory subgroup analyses of patients without a tBRCA mutation (non-nested, non-tBRCA subpopulations [LOH+, LOH-, LOH unknown]), the HR for OS was 1.084 (95% CI: 0.841, 1.396; median 32.2 months for rucaparib vs 38.3 months for placebo). The median survival follow-up for all patients was 77 months (6.4 years) with a range of 2 days to 93 months (7.6 years).
At the time of the final analysis, 89% of patients in the placebo arm had received at least one subsequent treatment, of whom 46% received a PARP inhibitor. In the rucaparib arm, 78% of patients had received at least one subsequent treatment.
Cardiac electrophysiology
Concentration-QTcF prolongation analysis was conducted using data from 54 patients with a solid tumour administered continuous rucaparib at doses ranging from 40 mg once daily to 840 mg twice daily (1.4 times the approved recommended dose). At the predicted median steady-state Cmax following 600 mg rucaparib twice daily, the projected QTcF increase from baseline was 11.5 msec (90% CI: 8.77 to 14.2 msec). Thus, the risk for clinically significant QTcF increase from baseline (i.e. >20 msec) is low.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with Rubraca in all subsets of the paediatric population in ovarian cancer (see section 4.2 for information on paediatric use).
Pharmacokinetic properties
Plasma exposures of rucaparib, as measured by Cmax and AUC, were approximately dose proportional at evaluated doses (40 to 500 mg daily, 240 to 840 mg twice a day). Steady state was achieved after 1 week of dosing. Following repeated twice daily dosing, the accumulation based on AUC ranged from 3.5 to 6.2 fold.
Absorption
In patients with cancer following rucaparib 600 mg taken twice daily, the mean steady-state Cmax was 1940 ng/mL and AUC0-12h was 16900 h⋅ng/mL with Tmax of 1.9 hours. The mean absolute oral bioavailability following a single oral dose of 12 to 120 mg rucaparib was 36%. The absolute oral bioavailability at 600 mg has not been determined. In patients with cancer following a high-fat meal, the Cmax increased by 20%, the AUC0-24h increased by 38%, and the Tmax was delayed by 2.5 hours, as compared with dosing under fasted conditions. The food effect on PK was not considered clinically significant. Rubraca can be administered with or without food.
Distribution
The in vitro protein binding of rucaparib is 70.2% in human plasma at therapeutic concentration levels. Rucaparib preferentially distributed to red blood cells with a blood-to-plasma concentration ratio of 1.83. In patients with cancer, rucaparib had a steady-state volume of distribution of 113 L to 262 L following a single intravenous dose of 12 mg to 40 mg rucaparib.
Biotransformation
In vitro, rucaparib is metabolised primarily by CYP2D6, and to a lesser extent by CYP1A2, and CYP3A4. In a population PK analysis, no clinically relevant PK differences were observed among patients with different CYP2D6 phenotypes (including poor metabolizers, n=9; intermediate metabolizers, n=71; normal metabolizers, n=76; and ultra-rapid metabolizers, n=4) or patients with different CYP1A2 phenotypes (including normal metabolizers, n=28; hyperinducers, n=136). The results should be interpreted with caution due to the limited representation of some subgroup phenotypes.
Following administration of a single oral dose of [14C]-rucaparib to patients with solid tumours, unchanged rucaparib accounted for 64.0% of the radioactivity in plasma. Oxidation, N-demethylation, N-methylation, glucuronidation, and N-formylation were the major metabolic pathways for rucaparib. The most abundant metabolite was M324, an oxidative deamination product of rucaparib, accounting for 18.6% of the radioactivity in plasma. In vitro, M324 was at least 30 fold less potent than rucaparib against PARP-1, PARP-2, and PARP-3. Other minor metabolites accounted for 13.8% of the radioactivity in plasma. Rucaparib accounted for 44.9% and 94.9% of radioactivity in urine and faeces, respectively; while M324 accounted for 50.0% and 5.1% of radioactivity in urine and faeces, respectively.
Elimination
The clearance ranged from 13.9 to 18.4 L/hour, following a single intravenous dose of rucaparib 12 mg to 40 mg. Following administration of a single oral dose of [14C]-rucaparib 600 mg to patients, the overall mean recovery of radioactivity was 89.3%, with a mean recovery of 71.9% in faeces and 17.4% in urine by 288 hours post dose. Ninety percent of the observed faecal recovery was achieved within 168 hours postdose. The mean half-life (t ½) of rucaparib was 25.9 hours.
Medicinal product interactions
In vitro, rucaparib was shown to be a substrate of P-gp and BCRP, but not a substrate of renal uptake transporters OAT1, OAT3, and OCT2, or hepatic transporters OAPT1B1 and OATP1B3. Effect of P-gp and BCRP inhibitors on rucaparib PK cannot be ruled out.
In vitro, rucaparib reversibly inhibited CYP1A2, CYP2C19, CYP2C9, and CYP3A, and to a lesser extent CYP2C8, CYP2D6, and UGT1A1. Rucaparib induced CYP1A2, and down regulated CYP2B6 and CYP3A4 in human hepatocytes at clinically relevant exposures.
In vitro, rucaparib is a potent inhibitor of MATE1 and MATE2-K, a moderate inhibitor of OCT1, and a weak inhibitor of OCT2. At clinical exposures, rucaparib did not inhibit bile salt export pump (BSEP), OATP1B1, OATP1B3, OAT1 and OAT3. Inhibition of MRP4 by rucaparib cannot be fully ruled out at clinical exposures. No interaction with MRP2 or MRP3 was observed in vitro at the clinical exposure of rucaparib, however, mild bi-phasic activation and inhibition of MRP2 and concentration dependent inhibition of MRP3 were observed at concentrations higher than the observed plasma Cmax of rucaparib. The clinical relevance of MRP2 and MRP3 interaction in the gut is not known. In vitro, rucaparib is an inhibitor of the BCRP and P-gp efflux transporters. No significant P-gp inhibition was observed in vivo (section 4.5).
Population PK analysis suggested that concomitant use of PPIs is unlikely to have clinically meaningful impact on rucaparib PK. A firm conclusion cannot be made regarding the effect of co-administration of rucaparib and PPIs because dose level and time of administration have not been documented in detail for the PPIs.
Pharmacokinetics in specific populations
Age, race, and body weight
Based on population PK analysis, no clinically significant relationships were identified between predicted steady-state exposure and patient's age, race, and body weight. Patients included in the population PK study were aged 21 to 86 years (58% <65 years, 31% 65-74 years, and 11% >75 years), 82% were Caucasian, and had body weights between 41 and 171 kg (73% had body weight >60 kg).
Hepatic impairment
A population PK analysis was performed to evaluate the effect of hepatic impairment on the clearance of rucaparib in patients receiving rucaparib 600 mg twice daily. No clinically important differences were observed between 34 patients with mild hepatic impairment (total bilirubin ≤ ULN and AST > ULN or total bilirubin >1.0 to 1.5 times ULN and any AST) and 337 patients with normal hepatic function. In a study evaluating the pharmacokinetics of rucaparib in patients with hepatic impairment, patients with moderate hepatic impairment (N=8, National Cancer Institute - Organ Dysfunction Working Group criteria; total bilirubin >1.5 - ≤3 times ULN) had a 45% higher AUC of rucaparib following a single dose of 600 mg compared to patients with normal hepatic function (N=8). Cmax or Tmax were similar between the groups. No data are available for patients with severe hepatic impairment (see section 4.2).
Renal impairment
No formal studies of rucaparib in patients with renal impairment have been conducted. A population PK analysis was performed to evaluate the effect of renal impairment on the clearance of rucaparib in patients receiving rucaparib 600 mg twice daily. Patients with mild renal impairment (N=149; CLcr between 60 and 89 mL/min, as estimated by the Cockcroft-Gault method) and those with moderate renal impairment (N=76; CLcr between 30 and 59 mL/min) showed approximately 15% and 33% higher steady-state AUC, respectively, compared to patients with normal renal function (N=147; CLcr greater than or equal to 90 mL/min). The pharmacokinetic characteristics of rucaparib in patients with CLcr less than 30 mL/min or patients on dialysis are unknown (see section 4.2).
Preclinical safety data
General toxicology
The findings in non-clinical toxicology studies performed with oral rucaparib were generally consistent with the adverse events observed in clinical studies. In repeat-dose toxicity studies of up to 3 months duration in rats and dogs, the target organs were the gastrointestinal, haematopoietic, and lymphopoietic systems. These findings occurred at exposures below those observed in patients treated at the recommended dose, and were largely reversible within 4 weeks of cessation of dosing. In vitro, the IC50 of rucaparib against the human ether-à-go-go related gene (hERG) was 22.6 μM, which is approximately 13-fold higher than the Cmax in patients at the recommended dose.
Intravenous administration of rucaparib in the rat and dog induced cardiac effects at a high Cmax (5.4 to 7.3-fold higher than patients), but not at a lower Cmax (1.3 to 3.8-fold higher than patients). No cardiac effects were observed with oral dosing of rucaparib in repeat-dose toxicology studies at a rucaparib Cmax comparable to that observed in patients. Although no cardiac effects were observed following oral dosing, based on the findings in the intravenous studies and safety margins, cardiac effects in patients cannot be excluded when rucaparib is given orally.
Carcinogenicity
Carcinogenicity studies have not been performed with rucaparib.
Genotoxicity
Rucaparib was not mutagenic in a bacterial reverse mutation (Ames) assay. Rucaparib induced structural chromosomal aberrations in the in vitro human lymphocyte chromosomal aberration assay.
Reproductive toxicology
In an embryo-foetal development study in rats, rucaparib was associated with post-implantation loss at exposures of approximately 0.04 times the human AUC at the recommended dose.
Fertility studies have not been conducted with rucaparib. No effects on male and female fertility were observed in 3-month general toxicology studies in rats and dogs at exposures of 0.09 to 0.3 times the human AUC at the recommended dose. A potential risk cannot be ruled out based on the safety margin observed. In addition, according to its mechanism of action rucaparib may have the potential to impair fertility in humans.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.