RYBREVANT Concentrate for solution for infusion Ref.[28354] Active ingredients: Amivantamab
Source: European Medicines Agency (EU) Revision Year: 2025 Publisher: Janssen-Cilag International NV, Turnhoutseweg 30, B-2340 Beerse, Belgium
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Monoclonal antibodies and antibody drug conjugates
ATC code: L01FX18
Mechanism of action
Amivantamab is a low-fucose, fully-human IgG1-based EGFR-MET bispecific antibody with immune cell-directing activity that targets tumours with activating EGFR mutations such as Exon 19 deletions, Exon 21 L858R substitution, and Exon 20 insertion mutations. Amivantamab binds to the extracellular domains of EGFR and MET.
Amivantamab disrupts EGFR and MET signalling functions through blocking ligand binding and enhancing degradation of EGFR and MET, thereby preventing tumour growth and progression. The presence of EGFR and MET on the surface of tumour cells also allows for targeting of these cells for destruction by immune effector cells, such as natural killer cells and macrophages, through antibody-dependent cellular cytotoxicity (ADCC) and trogocytosis mechanisms, respectively.
Pharmacodynamic effects
Albumin
Amivantamab decreased serum albumin concentration, a pharmacodynamic effect of MET inhibition, typically during the first 8 weeks (see section 4.8); thereafter, albumin concentration stabilised for the remainder of amivantamab treatment.
Clinical efficacy and safety
Previously-untreated NSCLC with EGFR Exon 19 deletions or Exon 21 L858R substitution mutations (MARIPOSA)
NSC3003 (MARIPOSA) is a randomised, open-label, active-controlled, multicenter phase 3 study assessing the efficacy and safety of Rybrevant in combination with lazertinib as compared to osimertinib monotherapy as first-line treatment in patients with EGFR-mutated locally advanced or metastatic NSCLC not amenable to curative therapy. Patient samples were required to have one of the two common EGFR mutations (Exon 19 deletion or Exon 21 L858R substitution mutation), as identified by local testing. Tumour tissue (94%) and/or plasma (6%) samples for all patients were tested locally to determine EGFR Exon 19 deletion and/or Exon 21 L858R substitution mutation status using polymerase chain reaction (PCR) in 65% and next generation sequencing (NGS) in 35% of patients.
A total of 1074 patients were randomised (2:2:1) to receive Rybrevant in combination with lazertinib, osimertinib monotherapy, or lazertinib monotherapy until disease progression or unacceptable toxicity. Rybrevant was administered intravenously at 1050 mg (for patients <80 kg) or 1400 mg (for patients ≥80 kg) once weekly for 4 weeks, then every 2 weeks thereafter starting at week 5. Lazertinib was administered at 240 mg orally once daily. Osimertinib was administered at a dose of 80 mg orally once daily. Randomisation was stratified by EGFR mutation type (Exon 19 deletion or Exon 21 L858R), race (Asian or non-Asian), and history of brain metastasis (yes or no).
Baseline demographics and disease characteristics were balanced across the treatment arms. The median age was 63 (range: 25–88) years with 45% of patients ≥65 years; 62% were female; and 59% were Asian, and 38% were White. Baseline Eastern Cooperative Oncology Group (ECOG) performance status was 0 (34%) or 1 (66%); 69% never smoked; 41% had prior brain metastases; and 90% had Stage IV cancer at initial diagnosis. With regard to EGFR mutation status, 60% were Exon 19 deletions and 40% were Exon 21 L858R substitution mutations.
Rybrevant in combination with lazertinib demonstrated a statistically significant improvement in progression-free survival (PFS) by BICR assessment.
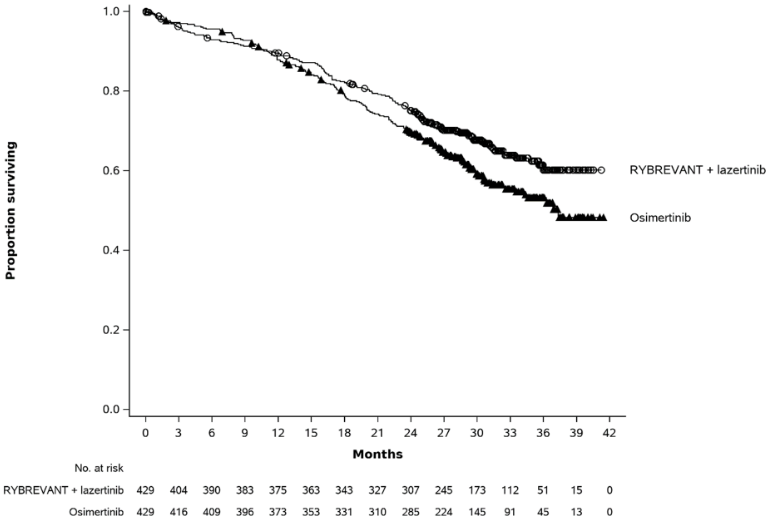
The final analysis of OS demonstrated a statistically significant improvement in OS for Rybrevant in combination with lazertinib compared to osimertinib (see Table 10 and Figure 2).
Table 10. Efficacy results in MARIPOSA:
| Rybrevant + lazertinib (N=429) | Osimertinib (N=429) | |
|---|---|---|
| Progression-free survival (PFS)a | ||
| Number of events | 192 (45%) | 252 (59%) |
| Median, months (95% CI) | 23.7 (19.1, 27.7) | 16.6 (14.8, 18.5) |
| Hazard Ratio (95% CI); p-value | 0.70 (0.58, 0.85); p=0.0002 | |
| Overall survival (OS) | ||
| Number of events | 173 (40%) | 217 (51%) |
| Median, months (95% CI) | NE (42.9, NE) | 36.7 (33.4, 41.0) |
| Hazard Ratio (95% CI); p-valueb | 0.75 (0.61, 0.92); p=0.0048 | |
| Objective response rate (ORR)a,b | ||
| ORR % (95% CI) | 80% (76%, 84%) | 77% (72%, 81%) |
| Duration of response (DOR)a,b | ||
| Median (95% CI), months | 25.8 (20.3, 33.9) | 18.1 (14.8, 20.1) |
BICR = blinded independent central review; CI = confidence interval; NE = not estimable PFS results are from data cut-off 11 August 2023 with a median follow-up of 22.0 months. DOR and ORR results are from data cut-off 13 May 2024 with a median follow-up of 31.3 months. OS results are from data cut-off 04 December 2024 with a median follow-up of 37.8 months.
a BICR by RECIST v1.1.
b Based on confirmed responders.
Figure 1. Kaplan-Meier curve of PFS in previously untreated NSCLC patients by BICR assessment:
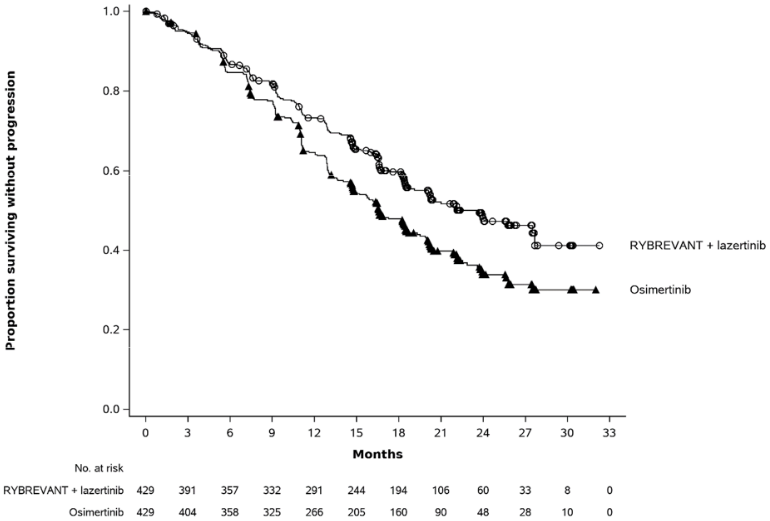
Figure 2. Kaplan-Meier curve of OS in previously untreated NSCLC patients:
Intracranial ORR and DOR by BICR were pre-specified endpoints in MARIPOSA. In the subset of patients with intracranial lesions at baseline, the combination of Rybrevant and lazertinib, demonstrated similar intracranial ORR to the control. Per protocol, all patients in MARIPOSA had serial brain MRIs to assess intracranial response and duration. Results are summarised in Table 11.
Table 11. Intracranial ORR and DOR by BICR assessment in subjects with intracranial lesions at baseline - MARIPOSA:
| Rybrevant + lazertinib (N=180) | Osimertinib (N=186) | |
|---|---|---|
| Intracranial Tumour Response Assessment | ||
| Intracranial ORR (CR+PR), % (95% CI) | 78% (71%, 84%) | 77% (71%, 83%) |
| Complete response | 64% | 59% |
| Intracranial DOR | ||
| Number of responders | 140 | 144 |
| Median, months (95% CI) | 35.0 (20.4, NE) | 25.1 (22.1, 31.2) |
CI = confidence interval
NE = not estimable
Intracranial ORR and DOR results are from data cut-off 04 Dec 2024 with a median follow-up of 37.8 months.
Previously treated NSCLC with EGFR Exon 19 deletions or Exon 21 L858R substitution mutations (MARIPOSA-2)
MARIPOSA-2 is a randomised (2:2:1) open-label, multicentre Phase 3 study in patients with locally advanced or metastatic NSCLC with EGFR Exon 19 deletions or Exon 21 L858R substitution mutations (mutation testing could have been performed at or after the time of locally advanced or metastatic disease diagnosis. Testing did not need to be repeated at the time of study entry once EGFR mutation status was previously established) after failure of prior therapy including a third-generation EGFR tyrosine kinase inhibitor (TKI). A total of 657 patients were randomised in the study, of which 263 received carboplatin and pemetrexed (CP); and 131 which received Rybrevant in combination with carboplatin and pemetrexed (Rybrevant-CP). Additionally, 236 patients were randomised to receive Rybrevant in combination with lazertinib, carboplatin, and pemetrexed in a separate arm of the study. Rybrevant was administered intravenously at 1400 mg (for patients <80 kg) or 1750 mg (for patients ≥80 kg) once weekly through 4 weeks, then every 3 weeks with a dose of 1750 mg (for patients <80 kg) or 2100 mg (for patients ≥80 kg) starting at Week 7 until disease progression or unacceptable toxicity. Carboplatin was administered intravenously at area under the concentration-time curve 5 mg/mL per minute (AUC 5) once every 3 weeks, for up to 12 weeks. Pemetrexed was administered intravenously at 500 mg/m² on once every 3 weeks until disease progression or unacceptable toxicity.
Patients were stratified by osimertinib line of therapy (first-line or second-line), prior brain metastases (yes or no), and Asian race (yes or no).
Of the 394 patients randomised to the Rybrevant-CP arm or CP arm, the median age was 62 (range: 31-85) years, with 38% of the patients ≥65 years of age; 60% were female; and 48% were Asian and 46% were White. Baseline Eastern Cooperative Oncology Group (ECOG) performance status was 0 (40%) or 1 (60%); 66% never smoked; 45% had history of brain metastasis, and 92% had Stage IV cancer at initial diagnosis.
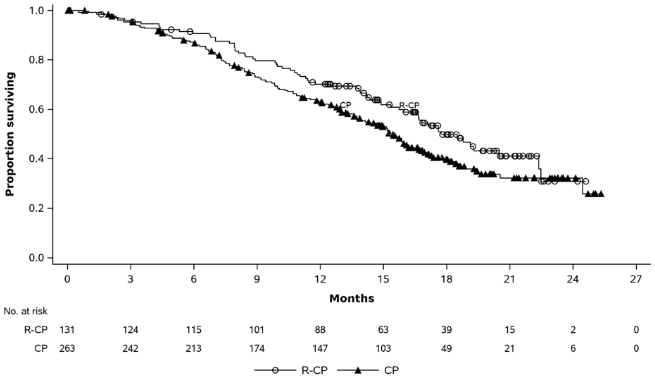
Rybrevant in combination with carboplatin and pemetrexed demonstrated a statistically significant improvement in progression-free survival (PFS) compared to carboplatin and pemetrexed, with a HR of 0.48 (95% CI: 0.36, 0.64; p<0.0001). At the time of the second interim analysis for OS, with a median follow-up of approximately 18.6 months for Rybrevant-CP and approximately 17.8 months for CP, the OS HR was 0.73 (95% CI: 0.54, 0.99; p=0.0386). This was not statistically significant (tested at a prespecified significance level of 0.0142).
Efficacy results are summarised in Table 12.
Table 12. Efficacy results in MARIPOSA-2:
| Rybrevant+ carboplatin+ pemetrexed (N=131) | carboplatin+ pemetrexed (N=263) | |
|---|---|---|
| Progression-free survival (PFS)a | ||
| Number of events (%) | 74 (57) | 171 (65) |
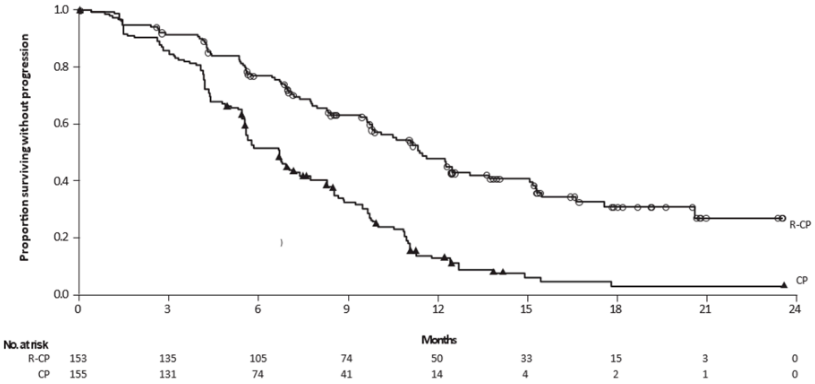
| Median, months (95% CI) | 6.3 (5.6, 8.4) | 4.2 (4.0, 4.4) |
| HR (95% CI); p-value | 0.48 (0.36, 0.64); p<0.0001 | |
| Overall survival (OS) | ||
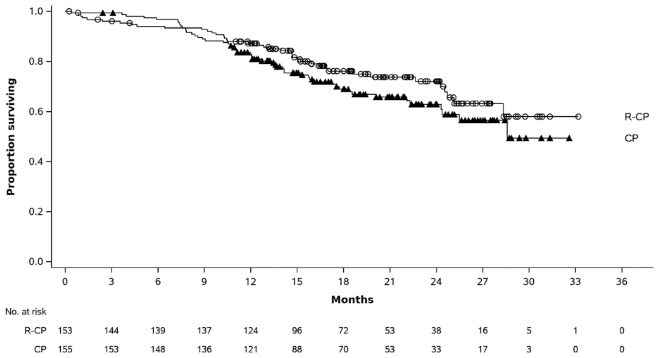
| Number of events (%) | 65 (50) | 143 (54) |
| Median, months (95% CI) | 17.7 (16.0, 22.4) | 15.3 (13.7, 16.8) |
| HR (95% CI); p-valueb | 0.73 (0.54, 0.99); p=0.0386 | |
| Objective response ratea | ||
| ORR, % (95% CI) | 64% (55%, 72%) | 36% (30%, 42%) |
| Odds Ratio (95% CI); p-value | 3.10 (2.00, 4.80); p<0.0001 | |
| Duration of response (DOR)a | ||
| Median (95% CI), months | 6.90 (5.52, NE) | 5.55 (4.17, 9.56) |
| Patients with DOR ≥6 months | 31.9% | 20.0% |
CI = Confidence Interval
PFS and ORR results are from data cut-off 10 July-2023 when hypothesis testing and final analysis for these endpoints was performed. OS results are from data cut-off 26 April 2024 from the second interim OS analysis.
a BICR-assessed
b The p-value is compared to a 2-sided significance level of 0.0142. Thus the OS results are not significant as of the second interim analysis.
Figure 3. Kaplan-Meier curve of PFS in previously treated NSCLC patients by BICR assessment:
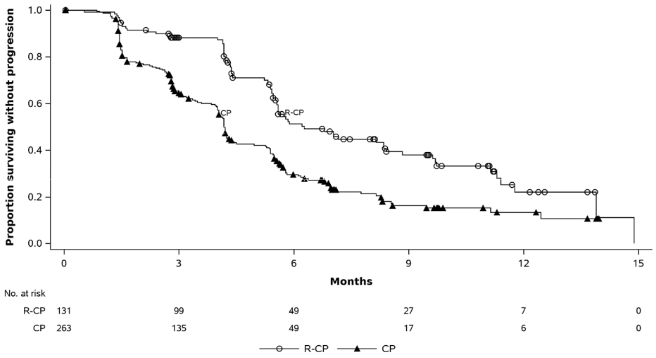
The PFS benefit of Rybrevant-CP compared to CP was consistent across all the predefined subgroups analysed, including ethnicity, age, gender, smoking history, and CNS metastases status at study entry.
Figure 4. Kaplan-Meier curve of OS in previously treated NSCLC patients:
Intracranial metastases efficacy data
Patients with asymptomatic or previously treated and stable intracranial metastases were eligible to be randomised in MARIPOSA-2. Treatment with Rybrevant-CP was associated with a numeric increase in intracranial ORR (23.3% for Rybrevant-CP versus 16.7% for CP, odds ratio of 1.52; 95% CI (0.51, 4.50), and intracranial DOR (13.3 months; 95% CI (1.4, NE) in the Rybrevant-CP arm compared with 2.2 months; 95% CI (1.4, NE) in the CP arm). The median follow-up for Rybrevant-CP was approximately 18.6 months.
Previously-untreated non-small cell lung cancer (NSCLC) with Exon 20 insertion mutations (PAPILLON)
PAPILLON is a randomised, open-label, multicentre Phase 3 study comparing treatment with Rybrevant in combination with carboplatin and pemetrexed to chemotherapy alone (carboplatin and pemetrexed) in patients with treatment-naïve, locally advanced or metastatic NSCLC with activating EGFR Exon 20 insertion mutations. Tumour tissue (92.2%) and/or plasma (7.8%) samples for all 308 patients were tested locally to determine EGFR Exon 20 insertion mutation status using next generation sequencing (NGS) in 55.5% of patients and/or polymerase chain reaction (PCR) in 44.5% of patients. Central testing was also performed using the AmoyDx LC10 tissue test, Thermo Fisher Oncomine Dx Target Test, and the Guardant 360 CDx plasma test.
Patients with brain metastases at screening were eligible for participation once they were definitively treated, clinically stable, asymptomatic, and off corticosteroid treatment for at least 2 weeks prior to randomisation.
Rybrevant was administered intravenously at 1400 mg (for patients <80 kg) or 1750 mg (for patients ≥80 kg) once weekly through 4 weeks, then every 3 weeks with a dose of 1750 mg (for patients <80 kg) or 2100 mg (for patients ≥80 kg) starting at Week 7 until disease progression or unacceptable toxicity. Carboplatin was administered intravenously at area under the concentration-time curve 5 mg/mL per minute (AUC 5) once every 3 weeks, for up to 12 weeks. Pemetrexed was administered intravenously at 500 mg/m² on once every 3 weeks until disease progression or unacceptable toxicity. Randomisation was stratified by ECOG performance status (0 or 1), and prior brain metastases (yes or no). Patients randomised to the carboplatin and pemetrexed arm who had confirmed disease progression were permitted to cross over to receive Rybrevant monotherapy. A total of 308 subjects were randomised (1:1) to Rybrevant in combination with carboplatin and pemetrexed (N=153) or carboplatin and pemetrexed (N=155). The median age was 62 (range: 27 to 92) years, with 39% of the subjects ≥65 years of age; 58% were female; and 61% were Asian and 36% were White. Baseline Eastern Cooperative Oncology Group (ECOG) performance status was 0 (35%) or 1 (64%); 58% never smoked; 23% had history of brain metastasis and 84% had Stage IV cancer at initial diagnosis.
The primary endpoint for PAPILLON was PFS, as assessed by BICR. The median follow-up was14.9 months (range: 0.3 to 27.0).
Efficacy results are summarised in Table 13.
Table 13. Efficacy results in PAPILLON:
| Rybrevant carboplatin pemetrexed (N=153) | carboplatin+ pemetrexed (N=155) | |
|---|---|---|
| Progression-free survival (PFS)a | ||
| Number of events | 84 (55%) | 132 (85%) |
| Median, months (95% CI) | 11.4 (9.8, 13.7) | 6.7 (5.6, 7.3) |
| HR (95% CI); p-value | 0.395 (0.29, 0.52); p<0.0001 | |
| Objective response ratea,b | ||
| ORR, % (95% CI) | 73% (65%, 80%) | 47% (39%, 56%) |
| Odds ratio (95% CI); p-value | 3.0 (1.8, 4.8); p<0.0001 | |
| Complete response | 3.9% | 0.7% |
| Partial response | 69% | 47% |
| Overall survival (OS)c | ||
| Number of events | 40 | 52 |
| Median OS, months (95% CI) | NE (28.3, NE) | 28.6 (24.4, NE) |
| HR (95% CI); p-value | 0.756 (0.50, 1.14); p=0.1825 | |
CI = confidence interval
NE = not estimable
a Blinded Independent Central Review by RECIST v1.1
b Based on Kaplan-Meier estimate.
c Based on the results of an updated OS with median follow-up of 20.9 months. The OS analysis was not adjusted for the potentially confounding effects of crossover (78 [50.3%] patients on the carboplatin + pemetrexed arm who received subsequent Rybrevant monotherapy treatment).
Figure 5. Kaplan-Meier curve of PFS in previously untreated NSCLC patients by BICR assessment:
The PFS benefit of Rybrevant in combination with carboplatin and pemetrexed compared to carboplatin and pemetrexed was consistent across all the predefined subgroups of brain metastases at study entry (yes or no), age (<65 or ≥65), sex (male or female), race (Asian or non-Asian), weight (<80 kg or ≥80 kg), ECOG performance status (0 or 1), and smoking history (yes or no).
Figure 6. Kaplan-Meier curve of OS in previously untreated NSCLC patients by BICR assessment:
Previously-treated non-small cell lung cancer(NSCLC) with Exon 20 insertion mutations (CHRYSALIS)
CHRYSALIS is a multicentre, open-label, multi-cohort study conducted to assess the safety and efficacy of Rybrevant in patients with locally advanced or metastatic NSCLC. Efficacy was evaluated in 114 patients with locally advanced or metastatic NSCLC who had EGFR Exon 20 insertion mutations, whose disease had progressed on or after platinum-based chemotherapy, and who had a median follow-up of 12.5 months. Tumour tissue (93%) and/or plasma (10%) samples for all patients were tested locally to determine EGFR Exon 20 insertion mutation status using next generation sequencing (NGS) in 46% of patients and/or polymerase chain reaction (PCR) in 41% of patients; for 4% of patients, the testing methods were not specified. Patients with untreated brain metastases or a history of ILD requiring treatment with prolonged steroids or other immunosuppressive agents within the last 2 years were not eligible for the study. Rybrevant was administered intravenously at 1050 mg for patients <80 kg or 1400 mg for patients ≥80 kg once weekly for 4 weeks, then every 2 weeks starting at Week 5 until loss of clinical benefit or unacceptable toxicity. The primary efficacy endpoint was investigator-assessed overall response rate (ORR), defined as confirmed complete response (CR) or partial response (PR) based on RECIST v1.1. In addition, the primary endpoint was assessed by a blinded independent central review (BICR). Secondary efficacy endpoints included duration of response (DOR).
The median age was 62 (range: 36–84) years, with 41% of the patients ≥65 years of age; 61% were female; and 52% were Asian and 37% were White. The median number of prior therapies was 2 (range: 1 to 7 therapies). At baseline, 29% had Eastern Cooperative Oncology Group (ECOG) performance status of 0 and 70% had ECOG performance status of 1; 57% never smoked; 100% had Stage IV cancer; and 25% had previous treatment for brain metastases. Insertions in Exon 20 were observed at 8 different residues; the most common residues were A767 (22%), S768 (16%), D770 (12%), and N771 (11%).
Efficacy results are summarised in Table 14.
Table 14. Efficacy results in CHRYSALIS:
| Investigator assessment (N=114) | |
|---|---|
| Overall response ratea,b (95% CI) | 37% (28%, 46%) |
| Complete response | 0% |
| Partial response | 37% |
| Duration of response | |
| Medianc (95% CI), months | 12.5 (6.5, 16.1) |
| Patients with DOR ≥6 months | 64% |
CI = Confidence Interval
a Confirmed response
b ORR and DOR results by investigator assessment were consistent with those reported by BICR assessment; ORR by BICR assessment was 43% (34%, 53%), with a 3% CR rate and a 40% PR rate, median DOR by BICR assessment was 10.8 months (95% CI: 6.9, 15.0), and patients with DOR ≥6 months by BICR assessment was 55%.
c Based on Kaplan-Meier estimate.
Anti-tumour activity was observed across studied mutation subtypes.
Elderly
No overall differences in effectiveness were observed between patients ≥65 years of age and patients <65 years of age.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with Rybrevant in all subsets of the paediatric population in non-small cell lung cancer (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Based on Rybrevant monotherapy data, amivantamab area under the concentration-time curve (AUC1week) increases proportionally over a dose range from 350 to 1750 mg.
Based on simulations from the population pharmacokinetic model, AUC1week was approximately 2.8-fold higher after the fifth dose for the 2-week dosing regimen and 2.6-fold higher after the fourth dose for the 3-week dosing regimen. Steady-state concentrations of amivantamab were reached by Week 13 for both the 3-week and 2-week dosing regimen and the systemic accumulation was 1.9-fold.
Distribution
Based on the individual amivantamab PK parameter estimates in population PK analysis, the geometric mean (CV%) total volume of distribution, is 5.12 (27.8%) L, following administration of the recommended dose of Rybrevant.
Elimination
Based on the individual amivantamab PK parameter estimates in population PK analysis, the geometric mean (CV%) linear clearance (CL) and terminal half-life associated with linear clearance is 0.266 (30.4%) L/day and 13.7 (31.9%) days respectively.
Special populations
Elderly
No clinically meaningful differences in the pharmacokinetics of amivantamab were observed based on age (27-87 years).
Renal impairment
No clinically meaningful effect on the pharmacokinetics of amivantamab was observed in patients with mild (60 ≤ creatinine clearance [CrCl] < 90 mL/min), moderate (29 ≤ CrCl < 60 mL/min) or severe (15 ≤ CrCl < 29 mL/min) renal impairment. Data in patients with severe renal impairment are limited (n=1), but there is no evidence to suggest that dose adjustment is required in these patients. The effect of end-stage renal disease (CrCl < 15 mL/min) on amivantamab pharmacokinetics is unknown.
Hepatic impairment
Changes in hepatic function are unlikely to have any effect on the elimination of amivantamab since IgG1-based molecules such as amivantamab are not metabolised through hepatic pathways.
No clinically meaningful effect in the pharmacokinetics of amivantamab was observed based on mild [(total bilirubin ≤ ULN and AST > ULN) or (ULN < total bilirubin ≤ 1.5 x ULN)] or moderate (1.5×ULN < total bilirubin ≤ 3×ULN and any AST) hepatic impairment. Data in patients with moderate hepatic impairment are limited (n=1), but there is no evidence to suggest that dose adjustment is required in these patients. The effect of severe (total bilirubin > 3 times ULN) hepatic impairment on amivantamab pharmacokinetics is unknown.
Paediatric population
The pharmacokinetics of Rybrevant in paediatric patients have not been investigated.
5.3. Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of repeated dose toxicity.
Carcinogenicity and mutagenicity
No animal studies have been performed to establish the carcinogenic potential of amivantamab. Routine genotoxicity and carcinogenicity studies are generally not applicable to biologic pharmaceuticals as large proteins cannot diffuse into cells and cannot interact with DNA or chromosomal material.
Reproductive toxicology
No animal studies have been conducted to evaluate the effects on reproduction and foetal development; however, based on its mechanism of action, amivantamab can cause foetal harm or developmental anomalies. As reported in the literature, reduction, elimination, or disruption of embryo foetal or maternal EGFR signaling can prevent implantation, cause embryo foetal loss during various stages of gestation (through effects on placental development), cause developmental anomalies in multiple organs or early death in surviving foetuses. Similarly, knock out of MET or its ligand hepatocyte growth factor (HGF) was embryonic lethal due to severe defects in placental development, and foetuses displayed defects in muscle development in multiple organs. Human IgG1 is known to cross the placenta; therefore, amivantamab has the potential to be transmitted from the mother to the developing foetus.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.