SANDOMIGRAN Tablet Ref.[27801] Active ingredients:
Source: Health Products and Food Branch (CA) Revision Year: 2012
Detailed pharmacology
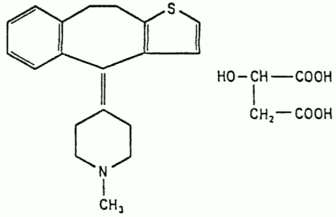
Pizotifen is a white to yellowish crystalline powder, readily soluble in water and organic solvents, and is designated chemically as 4 (9,10-Dihydro-4H-benzo[4,5] cycloheptal [1,2 b]thien-4-ylidene)-1-methylpiperidine hydrogen malate. It has the following structural formula:
A variety of in vitro and in vivo laboratory investigations indicated antagonistic or blocking actions of pizotifen towards serotonin and histamine and relatively weak anticholinergic activity. Pizotifen had very little effect as an epinephrine or bradykinin antagonist.
Tests for potentiation of barbiturate anesthesia and inhibition of locomotion in the mouse indicated that pizotifen had weak sedative properties. However, pizotifen was found to be more active in rats than either imipramine or amitriptyline in the antagonism of tetrabenazine-induced depression.
Pizotifen given orally (40 mg/kg), subcutaneously (5 mg/kg) and intravenously (1.25 mg/kg) to male Rhesus monkeys produced slight sedation, but no change in cardiac or respiratory rates during the subsequent four hours.
Pizotifen given intravenously (i.v.; 1 to 10 mg/kg) rapidly produced hypotension in dogs; reversion to normal pressures occurred within 30 minutes. Immediate increased heart rates were produced by the maximal dose but they quickly subsided. Blood pressure response to adrenaline was enhanced (2 mg/kg i.v.).
Blood sugar studies of normal and alloxan-treated rats did not indicate any hypoglycemic effects of pizotifen.
Pharmacokinetics
Absorption
Absorption half-life of pizotifen in man by the gastro-intestinal tract is 0.5 to 0.8 hours and nearly complete (80%). The absolute bioavailability is 78%. Maximum blood levels are reached 5 hours after oral administration.
Metabolism
Pizotifen is highly metabolised. Metabolism is mostly achieved through glucuronidation, and the main metabolite, N-glucuronide conjugate, accounts for at least 50% of the plasma and 60-70% of urinary excreted radioactivity.
Distribution
Plasma protein binding of pizotifen is over 90%. The distribution volume in man is 833L and 70L for pizotifen and N-glucuronide conjugate, respectively.
Elimination
Excretion of pizotifen by the feces is equivalent to about one-third of the given oral dose. Less than 1% of the administered dose is excreted unchanged in the urine, whereas up to 55% is excreted as metabolites. The elimination half-life for pizotifen and N-glucuronide conjugate is about 23 hours.
Toxicology
Acute Toxicity
Acute toxicity studies were conducted in mice, rats and rabbits:
| LD50 (mg/kg) | |||||
|---|---|---|---|---|---|
| Mouse | Rat | Rabbit | |||
| Oral | I.V. | Oral | I.V. | Oral | I.V. |
| 880 | 43 | 1500 | 17 | 700 | 19 |
| ±102 | ±2 | ±330 | ±1 | ±252 | ±2 |
Signs of toxicity after oral administration to mice and rats included motor disturbances (ataxia, jumping, twitching, sporadic convulsions, hyperreflexia), ventral decubitus, prostration, stupor, dyspnea and bradypnea. The signs lasted several hours in mice and up to 60 hours in rats. In rabbits, ventral decubitus, weakness and drooping of the head were observed. In all three species prior to death, ataxia, ventral or lateral decubitus, convulsions, dyspnea, paralysis and cyanosis were observed.
Chronic Toxicity
Chronic oral toxicity studies were done in both rats and dogs for durations of 26 weeks and two years.
Pizotifen was administered orally to rats at 3 dosage levels (5, 16 and 55 mg/kg/day) for 26 weeks. Weights of livers, adrenals and thyroids were increased in the high-dose group and there were mild signs of dose-dependent, centrolobular hepatic lipidosis and thyroidal hyperactivity in the mid and high-dose groups. There was no evidence of cholestasis.
Doses of 3, 9 and 27 mg/kg/day were administered to rats for 2 years. Cellular and clinical hematologic, ophthalmoscopic and histologic examinations did not indicate drug-induced abnormalities in animals sacrificed after 16 months. The only changes observed after 2 years of treatment were increased liver and kidney weights in high-dose females and increased liver weights in mid-dose females.
Pizotifen was administered to dogs at levels of 3, 10 and 30 mg/kg/day for 26 weeks. Increased relative organ weights of spleens, livers and thyroids were observed in the mid- and high-dose dogs. Microscopy indicated thyroidal hyperactivity and increased hepatic cellular turnover in a high-dose dog. Serum alkaline phosphatase was slightly increased in one mid-dose and one highdose dog at the end of the experiment. There was no evidence of cholestasis.
Doses of 1, 3 and 9 mg/kg/day also were administered to dogs for 2 years. Mean SGPT values were increased in high-dose dogs compared to control means. Other liver function tests were within normal range. The effect was more pronounced in males. Concomitant histopathologic changes were not evident.
Reproductive Studies
Pizotifen was administered orally at the dose levels of 3, 10, and 30 mg/kg/day to female rats and rabbits from the 6th to 15th, and 6th to 18th days of pregnancy respectively. The animals were sacrificed at term and examined together with their fetuses. Embryotoxic or teratogenic drug effects could not be detected in either animal species. Male and female fertility tests were done. Conception rates, litter sizes and litter weights were not affected.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.