SELADELPAR GILEAD Hard capsule Ref.[115056] Active ingredients: Seladelpar
Source: European Medicines Agency (EU) Revision Year: 2025 Publisher: Gilead Sciences Ireland UC, Carrigtohill, County Cork, T45 DP77, Ireland
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Bile and liver therapy, other drugs for bile therapy
ATC code: A05AX07
Mechanism of action
Seladelpar is a peroxisome proliferator-activated receptor delta (PPARδ) agonist, or delpar. PPARδ is a nuclear receptor expressed in the liver and other tissues. PPARδ activation reduces bile acid synthesis in the liver through Fibroblast Growth Factor 21 (FGF21)-dependent downregulation of CYP7A1, the key enzyme for the synthesis of bile acids from cholesterol and by decreasing cholesterol synthesis and absorption. These actions result in lower bile acid exposure in the liver and reduced circulating bile acid levels.
Pharmacodynamic effects
In clinical studies, reduction in ALP was observed within 1 week, continued to decrease through Month 3, and was sustained through Month 24.
In RESPONSE, treatment with seladelpar led to a decrease in Interleukin-31 (IL-31) after 6 and 12 months of treatment in patients with moderate to severe pruritus.
Clinical efficacy and safety
The efficacy of seladelpar was evaluated in patients with PBC in a randomised, double-blind, placebo-controlled, 12-month trial (RESPONSE). Patients were included in the trial if their ALP was 1.67-times upper limit of normal (ULN) or greater and total bilirubin was less than or equal to 2-times ULN. Patients were excluded from the trial if they had other chronic liver diseases, clinically important hepatic decompensation including portal hypertension with complications, or cirrhosis with complications (e.g., Model for End Stage Liver Disease [MELD] score of 12 or greater, known oesophageal varices or history of variceal bleeds, history of hepatorenal syndrome). A 14-day run-in period prior to randomisation was used to establish a baseline itch intensity, as measured by the patient-reported daily 24-hour Numerical Rating Scale (Pruritus-NRS) scores (0 “no itch” to 10 “worst itch imaginable”).
Patients were randomised (2:1) to receive either seladelpar (n=128) 10 mg once daily; or placebo (n=65) for 12 months. Seladelpar or placebo was administered in combination with UDCA in 181 (94%) patients during the trial, or as a monotherapy in 12 (6%) patients who were unable to tolerate UDCA.
The two treatment groups were generally balanced with respect to baseline demographics and disease characteristics. Of the 193 randomised patients, the mean age was 56.7 years (range 28-75 years); 41 (21%) were aged 65 years or older; 183 (95%) were female; 170 (88%) were White, 11 (6%) were Asian, 4 (2%) were Black or African American, 6 (3%) were American Indian or Alaska Native. A total of 56 (29%) patients identified as Hispanic/Latino.
The mean baseline ALP concentration was 314.3 U/L, corresponding to 2.7-times ULN. The mean baseline total bilirubin concentration was 0.758 mg/dL and was less than or equal to the ULN in 87% of the enrolled patients. At baseline patients in the study population had the following elevations in other liver biochemistries: alanine aminotransferase (ALT) 1.2-times the ULN, aspartate aminotransferase (AST) 1.2-times the ULN, and gamma-glutamyl transferase (GGT) 1.7-times the ULN. The baseline mean (SD) pruritus NRS score was 3.0 (2.85). Among enrolled patients, 49 (38%, mean NRS score 6.1) in the seladelpar 10 mg arm and 23 (35%, mean NRS score 6.6) in the placebo arm had moderate to severe pruritus (NRS score ≥ 4) at baseline (mean baseline NRS score 6.3).
Cirrhosis (Child-Pugh A) was present at baseline in 18 patients (14%) in the seladelpar 10 mg arm, and 9 patients (14%) in the placebo arm.
In RESPONSE, the primary endpoint was a responder analysis at Month 12, where response was defined as a composite of three criteria: ALP less than 1.67-times the ULN, total bilirubin ≤ ULN, and an ALP decrease of at least 15%. The ULN for ALP was defined as 116 U/L for females and males. The ULN for total bilirubin was defined as 1.1 mg/dL for females and males. ALP normalisation was defined as achieving ALP less than ≤1.0-times the ULN. The pruritus improvement was assessed by change from baseline in weekly averaged pruritus NRS score at Month 6 in patients with NRS score ≥4 at baseline.
The results for the primary composite endpoint and ALP normalisation are presented in Table 2.
Table 2. RESPONSE trial: Composite biochemical endpoint and ALP normalisation with seladelpar with or without UDCAa:
| Seladelpar 10 mg (N=128) | Placebo (N=65) | Treatment difference % (95% CI)e | |
|---|---|---|---|
| Primary composite endpoint at Month 12b | |||
| Responder rate, (%)c [95% CI] | 62 [53, 70] | 20 [10, 30] | 42 (28, 53) |
| Components of primary endpoint | |||
| ALP less than 1.67-times ULN, (%) | 66 | 26 | 39 (25, 52) |
| Decrease in ALP of at least 15%, (%) | 84 | 32 | 51 (37, 63) |
| Total bilirubin less than or equal to ULNd, (%) | 81 | 77 | 4 (-7, 17) |
| ALP normalisation | |||
| ALP normalisation at Month 12, ≤1.0×ULN (%)c [95% CI] | 25 [18, 33] | 0 [0, 0] | 25 (18, 33) |
N = Number
CI = Confidence Interval
a In the trial there were 12 patients (6%) who were intolerant to UDCA and initiated treatment as monotherapy: 8 subjects (6%) in the seladelpar 10 mg arm, and 4 patients (6 %) in the placebo arm.
^b6 Percentage of patients achieving a response, defined as an ALP value less than 1.67-times the ULN, an ALP decrease of at least 15%, and total bilirubin less than or equal to the ULN. Patients with missing values were considered as not achieving response.
c p<0.0001 for seladelpar 10 mg versus placebo. P-value was obtained using the Cochran–Mantel–Haenszel Test stratified by baseline ALP level <350 U/L versus ALP level ≥350 U/L, and baseline pruritus NRS <4 versus ≥4.
d The mean baseline total bilirubin value was 0.758 mg/dL and was less than or equal to the ULN in 87% of the enrolled patients.
e 95% unstratified Miettinen and Nurminen confidence intervals (CIs) are provided.
Alkaline phosphatase (ALP)
Figure 1 shows the mean reductions in ALP in seladelpar-treated patients compared to placebo. Reductions were observed at Month 1, continued through Month 6, and were sustained through Month 12.
Figure 1. Change from baseline in ALP over 12 months in RESPONSE by treatment arm with or without UDCAa:
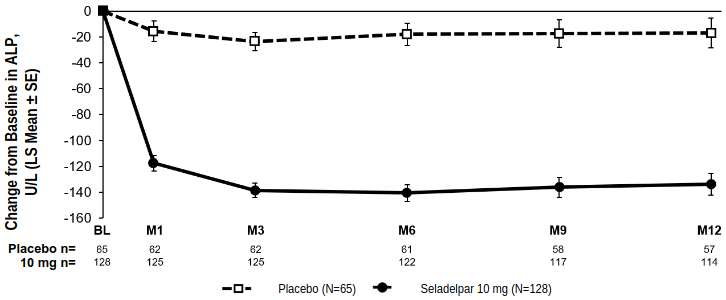
a In the trial there were 12 patients (6%) who were intolerant to UDCA and initiated treatment as monotherapy: 8 patients (6%) in the seladelpar 10 mg arm and 4 patients (6%) in the placebo arm.
Among the subset of patients with ALP <350 U/L (< approximately 3-times ULN) at baseline, 76% (71/93) and 23% (11/47) of patients achieved a response at Month 12, in the seladelpar 10 mg and placebo arms, respectively. For patients with ALP ≥350 U/L at baseline, 23% (8/35) and 11% (2/18) of patients achieved a response at Month 12, in the seladelpar 10 mg and placebo arms, respectively.
Lipid Parameters
The LS means difference from placebo in total cholesterol, LDL-C, and triglycerides was -4.4 (95% CI: -8.5, -0.3) mg/dL, -9.0 (95% CI: -15.0, -2.9) mg/dL, and -15.1 (95% CI: -22.1, -8.1), respectively at month 12. High density lipoprotein-cholesterol remained stable on treatment with seladelpar.
Pruritus
Seladelpar significantly reduced pruritus compared to placebo at Month 6 in patients with baseline average pruritus scores ≥4 as assessed by pruritus NRS score, a key secondary endpoint in the RESPONSE trial (Table 3). Seladelpar led to decreased patient-reported pruritus intensity by Month 1 which continued to decrease to Month 6.
Table 3. Change from Baseline in Pruritus Score at Month 6 in RESPONSE in PBC Patients with Moderate to Severe Pruritus at Baseline:
| Seladelpar 10 mg (N=49) | Placebo (N=23) | Treatment difference % (95% CI) | |
|---|---|---|---|
| Baseline average pruritus score, Mean (SD)b | 6.1 (1.4) | 6.6 (1.4) | - |
| Change from baseline in pruritus score at Month 6c | |||
| Mean (SE) | -3.2 (0.28) | -1.7 (0.41) | -1.5 (-2.5, -0.5)d |
a Assessed using the pruritus NRS, which evaluated patients' daily worst itching intensity on an 11-point rating scale with scores ranging from 0 (“no itching”) to 10 (“worst itching imaginable”). The pruritus NRS was administered daily in a ≥14-day run-in period prior to randomization through Month 6. Moderate to severe pruritus was defined as a pruritus NRS score ≥4.
b Baseline included mean of all daily recorded scores during the run in-period and on Day 1. The pruritus scores for each patient for post-baseline months were calculated by averaging the pruritus NRS scores within the scheduled week each month.
c Based on LS means from a mixed-effect model for repeated measures (MMRM) for change from baseline at Months 1 (Week 4), 3 (Week 12), and 6 (Week 26) accounting for baseline average pruritus score, baseline ALP level (<350 U/L versus ALP level ≥350 U/L), treatment arm, time (in months), and treatment-by-time interaction.
d p<0.005 for seladelpar 10 mg versus placebo.
The effect of seladelpar on pruritus was also assessed by additional patient-reported outcome measures in RESPONSE. At Month 6, an improvement in pruritus, as observed by reductions in total scores of the PBC-40 Itch Domain and 5-D Itch scale, was seen with seladelpar. (Table 4).
Table 4. Change from baseline in PBC-40 Itch Domain and 5-D Itch Scale total scores at Month 6 in RESPONSE in PBC patients with moderate to severe pruritus at baseline:
| Seladelpar 10 mg (N=49) | Placebo (N=23) | Treatment difference (95 CI) | |
|---|---|---|---|
| PBC-40 Itch Domaina | |||
| Mean (SE) | -2.2 (0.38) | -0.40 (0.60) | -1.8 (-3.2, -0.39) |
| 5-D Itch Scaleb | |||
| Mean (SE) | -4.7 (0.53) | -1.3 (0.80) | -3.4 (-5.3, -1.5) |
a LS means were obtained using MMRM for change from baseline at Month 6 accounting for baseline PBC-40 Quality of Life Itch Domain score, baseline ALP level (<350 U/L versus ALP level ≥350 U/L), treatment arm, time (in months), and treatment-by-time interaction.
b LS means were obtained using MMRM for change from baseline at Month 6 accounting for baseline 5-D Itch scale, baseline ALP level (<350 U/L versus ALP level ≥350 U/L), time (in months), treatment arm, and treatment-by-time interaction.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with seladelpar in all subsets of the paediatric population in treatment of primary biliary cholangitis (see section 4.2 for information on paediatric use).
This medicinal product has been authorised under a so-called ‘conditional approval’ scheme.
This means that further evidence on this medicinal product is awaited.
The European Medicines Agency will review any new information on this medicinal product at least every year and this SmPC will be updated as necessary.
5.2. Pharmacokinetic properties
Absorption
Following oral administration of a single dose of seladelpar 10 mg, seladelpar was readily absorbed with median time to peak concentration (tmax) of approximately 1.5 hours.
Seladelpar exposure increased approximately dose-proportionally with single doses from 2 mg to 15 mg, after which the increase in Cmax was larger than dose-proportional.
Seladelpar showed no evidence of meaningful drug accumulation after multiple daily dosing, and steady-state was achieved from day 4 onwards after daily dosing.
Co-administration of seladelpar with food delayed the tmax by 2.5 hours relative to fasted conditions and resulted in an approximately 32% reduction in the Cmax of seladelpar. As the overall exposure (AUC) is similar, the effects of food on seladelpar pharmacokinetics are not considered clinically relevant.
Distribution
In PBC patients, the steady state apparent volume of distribution of seladelpar is approximately 110.3 L. Plasma protein binding of seladelpar is greater than 99%.
Biotransformation
Seladelpar is primarily metabolized by CYP2C9 and to a lesser extent by CYP2C8 and CYP3A4. M2 is a major metabolite observed in human plasma, accounting for 17.6% of the total plasma radioactivity in the mass balance study and approximately doubling the plasma exposure compared to seladelpar. M2 is not expected to have clinically relevant pharmacological effects.
Elimination
In PBC patients, the apparent oral clearance of seladelpar is 12.6 L/h. Following administration of a single dose of 10 mg seladelpar in healthy subjects, mean elimination half-life was 6 hours for seladelpar. In PBC patients, the half-life range was 3.8 to 6.7 hours for seladelpar.
Following administration of an oral dose of radiolabelled seladelpar, 92.9% of radioactivity was recovered: 73.4% in urine and 19.5% in faeces. Urinary excretion of the dose as unchanged seladelpar was negligible (less than 0.01%).
Characteristics in specific groups or special populations
CYP2C9 genotype
Seladelpar is primarily metabolised in vitro by CYP2C9 which is a polymorphic enzyme. Seladelpar plasma exposure (dose-normalised AUC0-inf) was 18% higher in CYP2C9 intermediate metabolisers (*1/*2, *1/*8, *1/*3, *2/*2, n=28) compared to CYP2C9 normal metabolisers (*1/*1, n=84) after a single dose of seladelpar 1 mg to 15 mg. No conclusion could be made for poor metabolisers due to only one identified subject with *2/*3 and no subjects with *3/*3 were identified
Age, weight, gender and race
Based on population pharmacokinetic analysis, age (19 to 79 years old), weight (45.8 to 127.5 kg), gender, and race (White, Black, Asian, other) do not have a clinically meaningful effect on the pharmacokinetics of seladelpar. No dose adjustments are warranted based on these factors.
Renal impairment
In a dedicated clinical study of patients with mild (eGFR ≥60 to <90 mL/min), moderate (eGFR ≥30 to <60 mL/min), and severe (<30 mL/min and not on dialysis) renal impairment, the AUC0-inf of seladelpar was 48%, 33% and 3% greater than in patients with normal renal function, respectively, after administration of a single 10 mg dose of seladelpar. The Cmax of seladelpar was similar in patients with renal impairment, compared to patients with normal renal function. These differences in seladelpar AUC0-inf are not considered to be clinically meaningful. No dose adjustment of seladelpar is required for patients with mild, moderate, or severe renal impairment.
The pharmacokinetics of seladelpar have not been studied in patients requiring haemodialysis.
Hepatic impairment
Based on a clinical pharmacology study in subjects with mild, moderate, and severe hepatic impairment (Child-Pugh A, B, and C, respectively), seladelpar AUC was increased 1.10, 2.52, and 2.12-fold, and Cmax was increased 1.33, 5.19, and 5.03-fold, respectively, compared to subjects with normal hepatic function.
In an additional study, seladelpar exposures (Cmax, AUC) were 1.7 to 1.8-fold higher in PBC patients with mild hepatic impairment (Child-Pugh A) with portal hypertension and 1.6 to 1.9-fold higher in PBC patients with moderate hepatic impairment (Child-Pugh B), compared to PBC patients with mild hepatic impairment without portal hypertension, after a single oral dose of 10 mg seladelpar.
Following administration of 10 mg seladelpar once daily for 28 days in PBC patients with mild hepatic impairment (Child-Pugh A) with portal hypertension and PBC patients with moderate hepatic impairment (Child-Pugh B), there was no clinically meaningful accumulation of seladelpar (accumulation ratios were less than 1.2-fold).
Drug interaction studies
Effect of seladelpar on other medicinal products
Seladelpar has no clinically relevant effect on the pharmacokinetics of tolbutamide (CYP2C9 substrate), midazolam (CYP3A4 substrate), simvastatin (CYP3A4 and OATP substrate), atorvastatin (CYP3A4 and OATP substrate), and rosuvastatin (BCRP and OATP substrate).
Effect of other medicinal products on seladelpar
P-gp inhibitor:
In a dedicated clinical drug interaction study, seladelpar exposures were not significantly altered when a single dose of 600 mg quinidine (a P-gp inhibitor) was co-administered in healthy subjects.
5.3. Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential or toxicity to reproduction and development.
Reproductive and developmental toxicity
Seladelpar did not cause any foetal malformations or effects on embryo-foetal survival or growth in rats or rabbits. In rats, the exposure at NOAEL was 145-fold higher than the clinical AUC at the recommended dose of 10 mg, and 2-fold the clinical AUC in rabbits.
Oral administration of seladelpar at doses of 0, 5, 20 or 100 mg/kg/day in rats during gestation and lactation resulted in a dose-dependent reduction in pup body weights during the pre-weaning period at all dose levels, which was associated with slightly reduced pre-weaning survival at 100 mg/kg/day. Growth-related delays in developmental milestones were noted (eye opening and pinna unfolding at ≥5 mg/kg/day; hair growth and sexual maturity at 100 mg/kg/day). Growth reductions at 100 mg/kg/day continued into the post-weaning maturation period and were considered adverse. The exposure at the NOAEL of 20 mg/kg/day was 15-fold the clinical AUC.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.