SEPHIENCE Oral powder Ref.[115495] Active ingredients: Sepiapterin
Source: European Medicines Agency (EU) Revision Year: 2025 Publisher: PTC Therapeutics International Limited, Unit 1, 52-55 Sir John Rogerson's Quay, Dublin 2, D02 NA07, Ireland
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Other alimentary tract and metabolism products, Various alimentary tract and metabolism products
ATC code: A16AX28
Mechanism of action
Sepiapterin is a natural precursor of the enzymatic co-factor BH4, a critical co-factor for phenylalanine hydroxylase (PAH). Sepiapterin acts as a dual pharmacological chaperone (sepiapterin and BH4 each with its own binding affinity to variant PAH), including PAH variants commonly found in PKU and known to be insensitive to BH4, to improve the activity of the defective PAH enzyme, achieving a high concentration of BH4 intracellularly. By enhancing the conformational stability of misfolded PAH enzyme and increasing the intracellular concentrations of BH4, sepiapterin is able to effectively reduce blood Phe levels.
Clinical efficacy and safety
The efficacy of sepiapterin was evaluated in three clinical studies in patients with PKU.
Study 1 (PTC923-MD-003-PKU) was a 2-part, global, double-blind, randomised, placebo-controlled clinical study of 157 patients of all ages with PKU.
Part 1 of the study tested for responsiveness to sepiapterin, with 14 days of open-label treatment with sepiapterin followed by a minimum of 14 days of sepiapterin washout. Further, 73.1% (114/156) of study participants demonstrated a ≥15% reduction in blood Phe levels in response to sepiapterin. The dose of sepiapterin in patients ≥2 years of age was 60 mg/kg/day.
Subjects were instructed to continue their usual diet without modification. Patients ≥2 years of age who experienced a ≥15% reduction in blood Phe levels were classified as responsive and continued into Part 2 (n=110). After the washout period from Part 1, patients were randomised equally to either sepiapterin 20 mg/kg/day for Weeks 1 and 2, 40 mg/kg/day for Weeks 3 and 4, 60 mg/kg/day for Weeks 5 and 6 (n=56), or placebo (n=54) for 6 weeks. The primary efficacy was assessed by the mean change in blood Phe levels from baseline to Weeks 5 and 6 in the sepiapterin-treated group as compared to the mean change in the placebo group in patients who demonstrated a ≥ 30% reduction in blood Phe levels during Part 1. In Part 2, demographics were well balanced between the 2 treatment arms (Table 7). The median age at the time of informed consent was 14 years (range: 2-54), and participants, in terms of race, were predominantly white (91.8%). More than half (65.5%) of the 110 participants had PKU diagnosed at birth, and the majority (82.7%) had 'biochemically defined' non-classical PKU.
Table 7. Demographics and baseline characteristics:
| Participants in Part 1 only (n=47) | Randomised and treated participants in Part 2 | Overall treated participants (n=157) | |||
|---|---|---|---|---|---|
| Sepiapterin (n=56) | Placebo (n=54) | Overall (n=110) | |||
| Age (years) | |||||
| n | 47 | 56 | 54 | 110 | 157 |
| Mean (SD) | 18.4 (15.07) | 16.5 (11.12) | 18.4 (10.65) | 17.4 (10.88) | 17.7 (12.24) |
| Median (min, max) | 15.0 (1, 61) | 13.0 (2, 47) | 15.0 (4, 54) | 14.0 (2, 54) | 14.0 (1, 61) |
| Age category, n (%) | |||||
| ≥1 - <2 years | 3 (6.4) | 0 | 0 | 0 | 3 (1.9) |
| ≥2 - <6 years | 5 (10.6) | 7 (12.5) | 3 (5.6) | 10 (9.1) | 15 (9.6) |
| ≥6 - <12 years | 11 (23.4) | 17 (30.4) | 12 (22.2) | 29 (26.4) | 40 (25.5) |
| ≥12 - <18 years | 10 (21.3) | 14 (25.0) | 19 (35.2) | 33 (30.0) | 43 (27.4) |
| ≥18 years | 18 (38.3) | 18 (32.1) | 20 (37.0) | 38 (34.5) | 56 (35.7) |
SD, standard deviation
The difference between the 2 treatment groups was statistically significant (p<0.0001) (Table 8).
Table 8. Mean change in blood Phe levels from baseline to Week 5 and Week 6 in Part 2 (primary analysis set with Phe reduction from baseline ≥30% during Part 1):
| Sepiapterin (n=49) | Placebo (n=49) | Difference sepiapterin vs placebo | p value | |
|---|---|---|---|---|
| Baseline* | ||||
| Mean (SD) | 646.11 (253.007) | 654.04 (261.542) | ||
| Weeks 5 and 6** | ||||
| Mean (SD) | 236.04 (174.942) | 637.85 (259.886) | ||
| Mean change from baseline (μmol/L) | -410.07 (204.442) | -16.19 (198.642) | ||
| Mean percent change from baseline (%) | -62.8% | 1.4% | ||
| LS mean estimate for the mean change from baseline | ||||
| LS mean (SE) | -415.75 (24.066) | -19.88 (24.223) | -395.87 (33.848) | <0.0001 |
| 95% CI | (-463.52, -367.97) | (-67.97, 28.21) | (-463.07, -328.66) | |
CI, confidence interval; LS, least squares; MMRM, mixed model for repeated measures; Phe, phenylalanine; SD, standard deviation; SE, standard error
* Baseline is the average of Day -1 and Day 1 blood Phe levels in Part 2.
** Blood Phe concentrations were based on average values during Weeks 5 and 6.
LS means, standard errors, confidence intervals, and p values were from an MMRM with change in blood Phe from baseline to post-baseline assessments as the response variable, and fixed effects for treatment, baseline blood Phe, baseline Phe stratum, visit, and treatment-by-visit interaction.
Similar responses were observed in the population of patients with classical PKU (cPKU), with a 69% reduction in blood Phe at Week 6 in subjects receiving sepiapterin (n=6) versus an increase of 3% after placebo (n=9).
Study 2 (PKU-002) was a Phase 2, randomised, double-crossover, open-label, active-controlled, proof-of-concept clinical study of sepiapterin in patients with PKU.
The study consisted of 6 sequence groups of 4 patients per group for a total of 24 patients. Each sequence group was randomised to receive 7-day treatments of sepiapterin 60 mg/kg/day, sepiapterin 20 mg/kg/day, and sapropterin dihydrochloride 20 mg/kg/day, in random order followed by a 7-day washout after each treatment. Preliminary efficacy was assessed by the reduction in blood Phe concentrations. Results of the primary efficacy weekly mean analysis demonstrated that treatment with sepiapterin resulted in a decrease in blood Phe concentrations relative to baseline that was statistically significant for all treatments (n=24). A greater proportion of patients receiving sepiapterin treatment, regardless of dose, experienced plasma Phe reductions of at least 10%, 20%, and 30% compared with patients receiving sapropterin 20 mg/kg/day. More patients receiving sepiapterin 60 mg/kg/day achieved normalised plasma Phe concentrations (<120 μmol/L) and blood Phe within the target range (≤360 μmol/L) compared with sapropterin 20 mg/kg/day. In subjects with cPKU, treatment with sepiapterin (60 mg/kg/day) resulted in a significant decrease in blood Phe concentration relative to baseline.
Study 3 (PTC923-MD-004-PKU) is an ongoing, Phase 3, multicentre, open-label clinical study to assess the safety and dietary Phe tolerance during long-term treatment with sepiapterin in patients with PKU. One hundred sixty-nine (169) patients received treatment with sepiapterin 7.5 mg/kg/day in participants 0 to <6 months of age, 15 mg/kg/day in participants 6 to <12 months of age, 30 mg/kg/day in participants 12 months to <2 years of age, or 60 mg/kg/day in participants ≥2 years of age. Interim data indicate that daily sepiapterin administration is associated with an approximately 2.3-fold increase in mean daily Phe consumption (27.6 mg/kg/day at baseline versus 62.5 mg/kg/day at Week 26) while maintaining Phe levels <360 μmol/L. The majority of subjects reached at least a 15% (76.7% of participants) or 30% (67.4% of participants) reduction in blood Phe (Figure 1).
Figure 1. Mean (SD) dietary Phe consumption over time during dietary Phe tolerance assessment (dietary Phe tolerance analysis set):
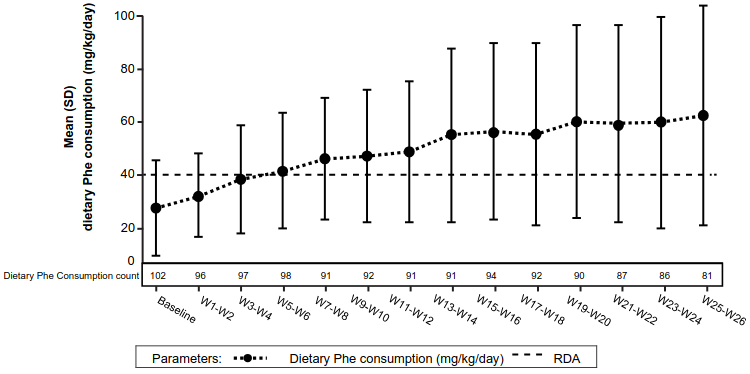
Phe, phenylalanine; PKU, phenylketonuria; RDA, recommended daily allowance; SD, standard deviation; W, week
Note: Baseline is defined as the average of daily dietary Phe consumption (mg/kg/day) at Month 1. The RDA is 0.8 g protein/kg, which is equivalent to approximately 40 mg/kg/day of Phe. Blood Phe levels baseline is the mean of the pre-assessment period Week 1–2. 1 g of protein is equivalent to approximately 50 mg of Phe.
These data indicate that sepiapterin treatment may allow liberalisation of the highly restrictive diet that patients with PKU must adhere to.
Subjects with history of allergies or adverse reactions to synthetic BH4 were excluded from the clinical studies.
The European Medicines Agency has deferred the obligation to submit the results of studies with Sephience in one or more subsets of the paediatric population in HPA (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Absorption
Following oral administration, sepiapterin is quickly absorbed, and the peak plasma concentrations occur in approximately 1 to 3 hours and decline to below the limit of quantitation (0.75 ng/mL) rapidly (generally by 12 hours). Maximum plasma sepiapterin concentration (Cmax) was approximately 2.80 ng/mL following the 60 mg/kg/day dose for 7 days with a high-fat high-calorie diet. No accumulation of sepiapterin was observed following repeated dosing.
Plasma sepiapterin is metabolised extensively to form the pharmacologically active metabolite BH4. The apparent terminal half-life for BH4 is approximately 5 hours. Both BH4 Cmax and area under the concentration-time curve from time zero to 24 hours postdose (AUC0-24h) increased with the dose, while the increase was less than dose proportional when the sepiapterin dose was above 20 mg/kg. There is no accumulation of BH4 following repeated doses of sepiapterin up to 60 mg/kg for 7 days.
Effect of food
When sepiapterin was administered with a low-fat, low-calorie meal in the dose range of 20 to 60 mg/kg, BH4 exposures were 1.69- to 1.72-fold higher for Cmax and 1.62- to 1.73-fold higher for AUC0-24h compared to administration under fasted conditions. When sepiapterin was administered with a high-fat, high-calorie meal, BH4 exposures were 2.21- to 2.26-fold higher for Cmax and 2.51- to 2.84-fold higher for AUC0-24h compared to administration under fasted conditions.
Sepiapterin can be taken with any meal at any time of the day at the same time every day.
Distribution
Binding of sepiapterin or BH4 to plasma protein is low, and the majority of sepiapterin and BH4 in plasma are free to exert pharmacological effects. In vitro studies show that sepiapterin is bound (mean 15.4%) to plasma protein in the presence of 0.1% dithiothreitol in the concentration range of 0.1 to 10 μM. BH4 was 41.3% (at 2 μM), 33.0% (at 5 μM), and 24.1% (at 15 μM) bound to protein in human plasma in the presence of 0.5% β-mercaptoethanol.
In healthy subjects, elevated BH4 concentration was observed in the cerebrospinal fluid following repeated sepiapterin oral administration.
Biotransformation
Sepiapterin is metabolised by SR/carbonyl reductase and DHFR in a 2-step unidirectional process to form BH4. The metabolism of BH4 is presumed to follow the same pathway as endogenous BH4, oxidised while acting as coenzymes for aromatic amino acid hydroxylases, such as PAH, tyrosine hydroxylase, tryptophan hydroxylase, and alkylglycerol monooxygenase, and nitric oxide synthase, and some metabolites, like 4α-hydroxy-tetrahydrobiopterin and quinonoid dihydrobiopterin, could be recycled to regenerate BH4 mediated by pterin-4α-carbinolamine dehydratase and dihydropteridine reductase.
Extensive metabolism of sepiapterin was observed in human following a single oral dose of 14C-sepiapterin. The major metabolic pathway involved oxidation/dehydrogenation, reduction/oxidation, oxidative deamination, dehydration, side chain cleavage, and methylation, etc, alone or in combination.
Elimination
Following oral administration in healthy human participants, sepiapterin was extensively metabolised with the metabolites excreted primarily in faeces. Plasma sepiapterin declined rapidly following Cmax to below the limit of quantitation, generally by 12 hours post-dose. Plasma BH4 declined mono-exponentially following Cmax. The terminal half-life was approximately 5 hours.
Following a single oral dose of 14C-sepiapterin to adult healthy subjects, a mean of 6.71% dosed radioactivity was recovered in urine and 26.18% in faeces with the combined total recovery of 32.9% by 240 hours. The majority of those radioactivity was recovered within the first 48 hours post-dose (28.2%). The total renal clearance of radioactivity derived from 14C-sepiapterin was 1.54 L/h (25.6 mL/min). Formation of volatile metabolites from sepiapterin in the gastrointestinal tract was confirmed in an in vitro study using human intestinal microbiota.
Special populations
Age
PKU patients of all ages had been included in the Phase 3 clinical studies. Except for allometric effect on clearance and volume of distribution, no further age effect was identified in the population PK study.
Ethnicity and race
Higher exposures to BH4 were observed for Asian subjects. In the Japanese ethno-bridging study, 10% to 24% higher AUC0-last and 14% to 29% higher Cmax of BH4 were observed in Japanese compared to non-Japanese subjects at sepiapterin dose range of 20 to 60 mg/kg.
Renal impairment
The PK and safety of sepiapterin have not been studied in patients with renal impairment.
Hepatic impairment
The PK and safety of sepiapterin have not been studied in patients with hepatic impairment.
Drug interactions
In vitro studies
In vitro studies indicate that sepiapterin and BH4 are unlikely to be perpetrators of CYP450-mediated metabolism.
In vitro, sepiapterin did not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, or CYP3A4, or induce CYP1A2, CYP2B6, or CYP3A4.
In vivo studies
In healthy subjects, co-administration of sepiapterin (20 mg/kg) with a single dose of the breast cancer resistance protein (BCRP) inhibitor curcumin (2 g) increased the exposures of BH4 slightly. The overall estimated geometric mean ratios (GMRs) (90% CI) for BH4 Cmax and area under the concentration-time curve from time zero to time of the last quantifiable measurement (AUC0-last) when sepiapterin was co-administered with curcumin compared to sepiapterin alone were 1.24 (1.15-1.33) and 1.20 (1.13 1.28), respectively. This modest increase is deemed not clinically relevant.
Co-administration of a single dose of sepiapterin at the maximum therapeutic dose of 60 mg/kg with the BCRP substrate rosuvastatin (10 mg) had no effect on the of rosuvastatin. The overall estimated GMRs (90% CI) for rosuvastatin Cmax and AUC0-last when rosuvastatin was co-administered with sepiapterin compared to rosuvastatin alone were 1.13 (1.00-1.28) and 1.02 (0.93-1.13), respectively.
5.3. Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, genotoxicity, carcinogenic potential and toxicity to reproduction and development.
In rats, following repeated oral administration, sepiapterin-related renal tubule degeneration/regeneration, interstitial inflammation, and fibrosis were noted as a result of crystal deposition in the papillary collecting tubules. These findings were partially reversible after a 4-week recovery period and no kidney toxicity occurred at BH4 exposure levels 2 times the clinical BH4 exposure levels at the maximum recommended human dose (MRHD).
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.