SEREVENT DISKUS Inhalation powder Ref.[10690] Active ingredients: Salmeterol
Source: FDA, National Drug Code (US) Revision Year: 2020
12.1. Mechanism of Action
Salmeterol is a selective LABA. In vitro studies show salmeterol to be at least 50 times more selective for beta2-adrenoceptors than albuterol. Although beta2-adrenoceptors are the predominant adrenergic receptors in bronchial smooth muscle and beta1-adrenoceptors are the predominant receptors in the heart, there are also beta2-adrenoceptors in the human heart comprising 10% to 50% of the total beta-adrenoceptors. The precise function of these receptors has not been established, but their presence raises the possibility that even selective beta2-agonists may have cardiac effects.
The pharmacologic effects of beta2-adrenoceptor agonist drugs, including salmeterol, are at least in part attributable to stimulation of intracellular adenyl cyclase, the enzyme that catalyzes the conversion of adenosine triphosphate (ATP) to cyclic-3′,5′-adenosine monophosphate (cyclic AMP). Increased cyclic AMP levels cause relaxation of bronchial smooth muscle and inhibition of release of mediators of immediate hypersensitivity from cells, especially from mast cells.
In vitro tests show that salmeterol is a potent and long-lasting inhibitor of the release of mast cell mediators, such as histamine, leukotrienes, and prostaglandin D2, from human lung. Salmeterol inhibits histamine-induced plasma protein extravasation and inhibits platelet-activating factor-induced eosinophil accumulation in the lungs of guinea pigs when administered by the inhaled route. In humans, single doses of salmeterol administered via inhalation aerosol attenuate allergen-induced bronchial hyper-responsiveness.
12.2. Pharmacodynamics
Inhaled salmeterol, like other beta-adrenergic agonist drugs, can produce dose-related cardiovascular effects and effects on blood glucose and/or serum potassium [see Warnings and Precautions (5.6, 5.10)]. The cardiovascular effects (heart rate, blood pressure) associated with salmeterol inhalation aerosol occur with similar frequency, and are of similar type and severity, as those noted following albuterol administration.
The effects of rising inhaled doses of salmeterol and standard inhaled doses of albuterol were studied in volunteers and in subjects with asthma. Salmeterol doses up to 84 mcg administered as inhalation aerosol resulted in heart rate increases of 3 to 16 beats/min, about the same as albuterol dosed at 180 mcg by inhalation aerosol (4 to 10 beats/min). Adult and adolescent subjects receiving 50-mcg doses of salmeterol inhalation powder (n=60) underwent continuous electrocardiographic monitoring during two 12-hour periods after the first dose and after 1 month of therapy, and no clinically significant dysrhythmias were noted. Also, pediatric patients receiving 50-mcg doses of salmeterol inhalation powder (n=67) underwent continuous electrocardiographic monitoring during two 12-hour periods after the first dose and after 3 months of therapy, and no clinically significant dysrhythmias were noted.
In 24-week clinical studies in patients with COPD, the incidence of clinically significant abnormalities on the predose ECGs at Weeks 12 and 24 in patients who received salmeterol 50 mcg was not different compared with placebo.
No effect of treatment with salmeterol 50 mcg was observed on pulse rate and systolic and diastolic blood pressure in a subset of patients with COPD who underwent 12-hour serial vital sign measurements after the first dose (n=91) and after 12 weeks of therapy (n=74). Median changes from baseline in pulse rate and systolic and diastolic blood pressure were similar for patients receiving either salmeterol or placebo [see Adverse Reactions (6.1)].
Concomitant Use of SEREVENT DISKUS with Other Respiratory Medications
Short-acting Beta2-agonists: In two 12-week repetitive-dose clinical trials in adult and adolescent subjects with asthma (N=149), the mean daily need for additional beta2-agonist in subjects using SEREVENT DISKUS was approximately 1½ inhalations/day. Twenty-six percent (26%) of the subjects in these trials used between 8 and 24 inhalations of short-acting beta-agonist per day on 1 or more occasions. Nine percent (9%) of the subjects in these trials averaged over 4 inhalations/day over the course of the 12-week trials. No increase in frequency of cardiovascular events was observed among the 3 subjects who averaged 8 to 11 inhalations/day; however, the safety of concomitant use of more than 8 inhalations/day of short-acting beta2-agonist with SEREVENT DISKUS has not been established. In 29 subjects who experienced worsening of asthma while receiving SEREVENT DISKUS during these trials, albuterol therapy administered via either nebulizer or inhalation aerosol (1 dose in most cases) led to improvement in FEV1 and no increase in occurrence of cardiovascular adverse events.
In 2 clinical trials in subjects with COPD, the mean daily need for additional beta2-agonist for subjects using SEREVENT DISKUS was approximately 4 inhalations/day. Twenty-four percent (24%) of subjects using SEREVENT DISKUS averaged 6 or more inhalations of albuterol per day over the course of the 24-week trials. No increase in frequency of cardiovascular adverse reactions was observed among subjects who averaged 6 or more inhalations per day.
Methylxanthines: The concurrent use of intravenously or orally administered methylxanthines (e.g., aminophylline, theophylline) by subjects receiving salmeterol has not been completely evaluated. In 1 clinical trial in subjects with asthma, 87 subjects receiving SEREVENT Inhalation Aerosol 42 mcg twice daily concurrently with a theophylline product had adverse event rates similar to those in 71 subjects receiving SEREVENT Inhalation Aerosol without theophylline. Resting heart rates were slightly higher in the subjects on theophylline but were little affected by therapy with SEREVENT Inhalation Aerosol.
In 2 clinical trials in subjects with COPD, 39 subjects receiving SEREVENT DISKUS concurrently with a theophylline product had adverse event rates similar to those in 302 subjects receiving SEREVENT DISKUS without theophylline. Based on the available data, the concomitant administration of methylxanthines with SEREVENT DISKUS did not alter the observed adverse event profile.
Cromoglycate: In clinical trials, inhaled cromolyn sodium did not alter the safety profile of salmeterol when administered concurrently.
12.3. Pharmacokinetics
Salmeterol xinafoate, an ionic salt, dissociates in solution so that the salmeterol and 1-hydroxy-2-naphthoic acid (xinafoate) moieties are absorbed, distributed, metabolized, and eliminated independently. Salmeterol acts locally in the lung; therefore, plasma levels do not predict therapeutic effect.
Absorption
Because of the small therapeutic dose, systemic levels of salmeterol are low or undetectable after inhalation of recommended doses (50 mcg of salmeterol inhalation powder twice daily). Following chronic administration of an inhaled dose of 50 mcg of salmeterol inhalation powder twice daily, salmeterol was detected in plasma within 5 to 45 minutes in 7 subjects with asthma; plasma concentrations were very low, with mean peak concentrations of 167 pg/mL at 20 minutes and no accumulation with repeated doses.
Distribution
The percentage of salmeterol bound to human plasma proteins averages 96% in vitro over the concentration range of 8 to 7,722 ng of salmeterol base per milliliter, much higher concentrations than those achieved following therapeutic doses of salmeterol.
Metabolism
Salmeterol base is extensively metabolized by hydroxylation, with subsequent elimination predominantly in the feces. No significant amount of unchanged salmeterol base was detected in either urine or feces.
An in vitro study using human liver microsomes showed that salmeterol is extensively metabolized to α-hydroxysalmeterol (aliphatic oxidation) by CYP3A4. Ketoconazole, a strong inhibitor of CYP3A4, essentially completely inhibited the formation of α-hydroxysalmeterol in vitro.
Elimination
In 2 healthy adult subjects who received 1 mg of radiolabeled salmeterol (as salmeterol xinafoate) orally, approximately 25% and 60% of the radiolabeled salmeterol was eliminated in urine and feces, respectively, over a period of 7 days. The terminal elimination half-life was about 5.5 hours (1 volunteer only).
The xinafoate moiety has no apparent pharmacologic activity. The xinafoate moiety is highly protein bound (>99%) and has a long elimination half-life of 11 days.
Drug Interaction Studies
Inhibitors of Cytochrome P450 3A4: Ketoconazole: In a placebo-controlled crossover drug interaction trial in 20 healthy male and female subjects, coadministration of salmeterol (50 mcg twice daily) and the strong CYP3A4 inhibitor ketoconazole (400 mg once daily) for 7 days resulted in a significant increase in plasma salmeterol exposure as determined by a 16-fold increase in AUC (ratio with and without ketoconazole 15.76 [90% CI: 10.66, 23.31]) mainly due to increased bioavailability of the swallowed portion of the dose. Peak plasma salmeterol concentrations were increased by 1.4-fold (90% CI: 1.23, 1.68). Three (3) out of 20 subjects (15%) were withdrawn from salmeterol and ketoconazole coadministration due to beta-agonist-mediated systemic effects (2 with QTc prolongation and 1 with palpitations and sinus tachycardia). Coadministration of salmeterol and ketoconazole did not result in a clinically significant effect on mean heart rate, mean blood potassium, or mean blood glucose. Although there was no statistical effect on the mean QTc, coadministration of salmeterol and ketoconazole was associated with more frequent increases in QTc duration compared with salmeterol and placebo administration.
Erythromycin: In a repeat-dose trial in 13 healthy subjects, concomitant administration of erythromycin (a moderate CYP3A4 inhibitor) and salmeterol inhalation aerosol resulted in a 40% increase in salmeterol Cmax at steady state (ratio with and without erythromycin 1.4 [90% CI: 0.96, 2.03], P = 0.12), a 3.6-beat/min increase in heart rate ([95% CI: 0.19, 7.03], P<0.04), a 5.8-msec increase in QTc interval ([95% CI: -6.14, 17.77], P = 0.34), and no change in plasma potassium.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
In an 18-month carcinogenicity study in CD-mice, salmeterol at oral doses of 1,400 mcg/kg and above (approximately 20 times the MRHDID for adults and children based on comparison of the plasma AUCs) caused a dose-related increase in the incidence of smooth muscle hyperplasia, cystic glandular hyperplasia, leiomyomas of the uterus, and ovarian cysts. No tumors were seen at 200 mcg/kg (approximately 3 times the MRHDID for adults and children based on comparison of the AUCs).
In a 24-month oral and inhalation carcinogenicity study in Sprague Dawley rats, salmeterol caused a dose-related increase in the incidence of mesovarian leiomyomas and ovarian cysts at doses of 680 mcg/kg and above (approximately 66 and 35 times the MRHDID for adults and children, respectively, on a mcg/m2 basis). No tumors were seen at 210 mcg/kg (approximately 20 and 10 times the MRHDID for adults and children, respectively, on a mcg/m2 basis). These findings in rodents are similar to those reported previously for other beta-adrenergic agonist drugs. The relevance of these findings to human use is unknown.
Salmeterol produced no detectable or reproducible increases in microbial and mammalian gene mutation in vitro. No clastogenic activity occurred in vitro in human lymphocytes or in vivo in a rat micronucleus test.
Fertility and reproductive performance were unaffected in male and female rats at oral doses up to 2,000 mcg/kg (approximately 195 times the MRHDID for adults on a mcg/m2 basis).
13.2. Animal Toxicology and/or Pharmacology
Preclinical
Studies in laboratory animals (minipigs, rodents, and dogs) have demonstrated the occurrence of cardiac arrhythmias and sudden death (with histologic evidence of myocardial necrosis) when beta-agonists and methylxanthines are administered concurrently. The clinical relevance of these findings is unknown.
14. Clinical Studies
14.1 Asthma
The initial trials supporting the approval of SEREVENT DISKUS for the treatment of asthma did not require the regular use of ICS. However, for the treatment of asthma, SEREVENT DISKUS is currently indicated only as concomitant therapy with an ICS [see Indications and Usage (1.1)].
Adult and Adolescent Subjects Aged 12 Years and Older
In 2 randomized double-blind trials, SEREVENT DISKUS was compared with albuterol inhalation aerosol and placebo in adolescent and adult subjects with mild-to-moderate asthma (protocol defined as 50% to 80% predicted FEV1, actual mean of 67.7% at baseline), including subjects who did and who did not receive concurrent ICS. The efficacy of SEREVENT DISKUS was demonstrated over the 12-week period with no change in effectiveness over this time period (Figure 1). There were no gender- or age-related differences in safety or efficacy. No development of tachyphylaxis to the bronchodilator effect was noted in these trials. FEV1 measurements (mean change from baseline) from these two 12-week trials are shown in Figure 1 for both the first and last treatment days.
Figure 1. Serial 12-Hour FEV1 from Two 12-Week Clinical Trials in Subjects with Asthma:
First Treatment Day:
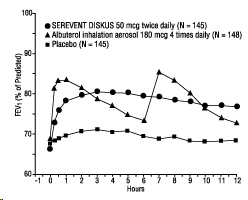
Last Treatment Day (Week 12):
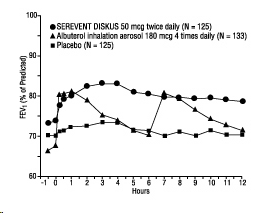
Table 4 shows the treatment effects seen during daily treatment with SEREVENT DISKUS for 12 weeks in adolescent and adult subjects with mild-to-moderate asthma.
Table 4. Daily Efficacy Measurements in Two 12-Week Clinical Trials (Combined Data)
| Parameter | Time | SEREVENT DISKUS | Albuterol Inhalation Aerosol | Placebo |
|---|---|---|---|---|
| No. of randomized subjects | 149 | 148 | 152 | |
| Mean AM peak expiratory flow (L/min) | Baseline 12 weeks | 395 | 394 | 394 |
| 427a | 394 | 396 | ||
| Mean % days with no asthma symptoms | Baseline 12 weeks | 13 | 12 | 14 |
| 33 | 21 | 20 | ||
| Mean % nights with no awakenings | Baseline 12 weeks | 63 | 68 | 70 |
| 85 a | 71 | 73 | ||
| Rescue medications (mean no. of inhalations per day) | Baseline 12 weeks | 4.3 | 4.3 | 4.2 |
| 1.6b | 2.2 | 3.3 | ||
| Asthma exacerbations (%) | 15 | 16 | 14 |
a Statistically superior to placebo and albuterol (P <0.001).
b Statistically superior to placebo (P <0.001).
Maintenance of efficacy for periods up to 1 year has been documented.
SEREVENT DISKUS and SEREVENT Inhalation Aerosol were compared with placebo in 2 additional randomized double-blind clinical trials in adolescent and adult subjects with mild-to-moderate asthma. SEREVENT DISKUS 50 mcg and SEREVENT Inhalation Aerosol 42 mcg, both administered twice daily, produced significant improvements in pulmonary function compared with placebo over the 12-week period. While no statistically significant differences were observed between the active treatments for any of the efficacy assessments or safety evaluations performed, there were some efficacy measures on which the metered-dose inhaler appeared to provide better results. Similar findings were noted in 2 randomized, single-dose, crossover comparisons of SEREVENT DISKUS and SEREVENT Inhalation Aerosol for the prevention of EIB. Therefore, while SEREVENT DISKUS was comparable to SEREVENT Inhalation Aerosol in clinical trials in mild-to-moderate subjects with asthma, it should not be assumed that they will produce clinically equivalent outcomes in all subjects.
Subjects on Concomitant Inhaled Corticosteroids: In 4 clinical trials in adult and adolescent subjects with asthma (N=1,922), the effect of adding SEREVENT Inhalation Aerosol to ICS therapy was evaluated over a 24-week treatment period. The trials compared the addition of salmeterol therapy to an increase (at least doubling) of the ICS dose.
Two randomized, double-blind, controlled, parallel-group clinical trials (N=997) enrolled subjects (aged 18 to 82 years) with persistent asthma who were previously maintained but not adequately controlled on ICS therapy. During the 2-week run-in period, all subjects were switched to beclomethasone dipropionate (BDP) 168 mcg twice daily. Subjects still not adequately controlled were randomized to either the addition of SEREVENT Inhalation Aerosol 42 mcg twice daily or an increase of BDP to 336 mcg twice daily. As compared with the doubled dose of BDP, the addition of SEREVENT Inhalation Aerosol resulted in statistically significantly greater improvements in pulmonary function and asthma symptoms, and statistically significantly greater reduction in supplemental albuterol use. The percent of subjects who experienced asthma exacerbations overall was not different between groups (i.e., 16.2% in the group receiving SEREVENT Inhalation Aerosol versus 17.9% in the higher-dose beclomethasone dipropionate group).
Two randomized, double-blind, controlled, parallel-group clinical trials (N=925) enrolled subjects (aged 12 to 78 years) with persistent asthma who were previously maintained but not adequately controlled on prior asthma therapy. During the 2- to 4-week run-in period, all subjects were switched to fluticasone propionate 88 mcg twice daily. Subjects still not adequately controlled were randomized to either the addition of SEREVENT Inhalation Aerosol 42 mcg twice daily or an increase of fluticasone propionate to 220 mcg twice daily. As compared with the increased (2.5 times) dose of fluticasone propionate, the addition of SEREVENT Inhalation Aerosol resulted in statistically significantly greater improvements in pulmonary function and asthma symptoms, and statistically significantly greater reductions in supplemental albuterol use. Fewer subjects receiving SEREVENT Inhalation Aerosol experienced asthma exacerbations than those receiving the higher dose of fluticasone propionate (8.8% versus 13.8%).
Table 5 shows the treatment effects seen during daily treatment with SEREVENT Inhalation Aerosol for 24 weeks in adolescent and adult subjects with mild-to-moderate asthma.
Onset of Action: During the initial treatment day in several multiple-dose clinical trials with SEREVENT DISKUS in subjects with asthma, the median time to onset of clinically significant bronchodilatation (≥15% improvement in FEV1) ranged from 30 to 48 minutes after a 50-mcg dose.
One hour after a single dose of 50 mcg of SEREVENT DISKUS, the majority of subjects had ≥15% improvement in FEV1. Maximum improvement in FEV1 generally occurred within 180 minutes, and clinically significant improvement continued for 12 hours in most subjects.
Pediatric Subjects
In a randomized, double-blind, controlled trial (N=449), 50 mcg of SEREVENT DISKUS was administered twice daily to pediatric subjects with asthma who did and who did not receive concurrent ICS. The efficacy of salmeterol inhalation powder was demonstrated over the 12-week treatment period with respect to periodic serial PEF (36% to 39% postdose increase from baseline) and FEV1 (32% to 33% postdose increase from baseline). Salmeterol was effective in demographic subgroup analyses (gender and age) and was effective when coadministered with other inhaled asthma medications such as short-acting bronchodilators and ICS. A second randomized, double-blind, placebo-controlled trial (N=207) with 50 mcg of salmeterol inhalation powder via an alternate device supported the findings of the trial with the DISKUS.
Salmeterol Multicenter Asthma Research Trial
The SMART trial was a randomized double-blind trial that enrolled LABA-naive subjects with asthma (average age of 39 years; 71% Caucasian, 18% African American, 8% Hispanic) to assess the safety of salmeterol (SEREVENT Inhalation Aerosol) 42 mcg twice daily over 28 weeks compared with placebo when added to usual asthma therapy.
A planned interim analysis was conducted when approximately half of the intended number of subjects had been enrolled (N=26,355), which led to premature termination of the trial. The results of the interim analysis showed that subjects receiving salmeterol were at increased risk for fatal asthma events (Table 5 and Figure 2). In the total population, a higher rate of asthma-related death occurred in subjects treated with salmeterol than those treated with placebo (0.10% versus 0.02%, relative risk: 4.37 [95% CI: 1.25, 15.34]).
Post hoc subpopulation analyses were performed. In Caucasians, asthma-related death occurred at a higher rate in subjects treated with salmeterol than in subjects treated with placebo (0.07% versus 0.01%, relative risk: 5.82 [95% CI: 0.70, 48.37]). In African Americans also, asthma-related death occurred at a higher rate in subjects treated with salmeterol than those treated with placebo (0.31% versus 0.04%, relative risk: 7.26 [95% CI: 0.89, 58.94]). Although the relative risks of asthma-related death were similar in Caucasians and African Americans, the estimate of excess deaths in subjects treated with salmeterol was greater in African Americans because there was a higher overall rate of asthma-related death in African American subjects (Table 5).
Post hoc analyses in pediatric subjects aged 12 to 18 years were also performed. Pediatric subjects accounted for approximately 12% of subjects in each treatment arm. Respiratory-related death or life-threatening experience occurred at a similar rate in the salmeterol group (0.12% [2/1,653]) and the placebo group (0.12% [2/1,622]; relative risk: 1.0 [95% CI: 0.1, 7.2]). All-cause hospitalization, however, was increased in the salmeterol group (2% [35/1,653]) versus the placebo group (less than 1% [16/1,622]; relative risk: 2.1 [95% CI: 1.1, 3.7]).
The data from the SMART trial were not adequate to determine whether concurrent use of ICS or other long-term asthma control therapy mitigated the risk of asthma-related death.
Table 5. Asthma-Related Deaths in the 28-Week Salmeterol Multicenter Asthma Research Trial (SMART):
| Salmeteroln (%a) | Placebon (%a) | Relative Riskb(95% Confidence Interval) | Excess Deaths Expressed per 10,000 Subjectsc(95% Confidence Interval) | |
|---|---|---|---|---|
| Total Populationd | ||||
| Salmeterol: n=13,176 | 13 (0.10%) | 4.37 (1.25, 15.34) | 8 (3, 13) | |
| Placebo: n=13,179 | 3 (0.02%) | |||
| Caucasian | ||||
| Salmeterol: n=9,281 | 6 (0.07%) | 5.82 (0.70, 48.37) | 6 (1, 10) | |
| Placebo: n=9,361 | 1 (0.01%) | |||
| African American | ||||
| Salmeterol: n=2,366 | 7 (0.31%) | 7.26 (0.89, 58.94) | 27 (8, 46) | |
| Placebo: n=2,319 | 1 (0.04%) | |||
a Life-table 28-week estimate, adjusted according to the subjects' actual lengths of exposure to study treatment to account for early withdrawal of subjects from the study.
b Relative risk is the ratio of the rate of asthma-related death in the salmeterol group and the rate in the placebo group. The relative risk indicates how many more times likely an asthma-related death occurred in the salmeterol group than in the placebo group in a 28-week treatment period.
c Estimate of the number of additional asthma-related deaths in subjects treated with salmeterol in SMART, assuming 10,000 subjects received salmeterol for a 28-week treatment period. Estimate calculated as the difference between the salmeterol and placebo groups in the rates of asthma-related death multiplied by 10,000.
d The Total Population includes the following ethnic origins listed on the case report form: Caucasian, African American, Hispanic, Asian, and "Other". In addition, the Total Population includes those subjects whose ethnic origin was not reported. The results for Caucasian and African American subpopulations are shown above. No asthma-related deaths occurred in the Hispanic (salmeterol n=996, placebo n=999), Asian (salmeterol n=173, placebo n=149), or "Other" (salmeterol n=230, placebo n=224) subpopulations. One asthma-related death occurred in the placebo group in the subpopulation whose ethnic origin was not reported (salmeterol n=130, placebo n=127).
Figure 2. Cumulative Incidence of Asthma-Related Deaths in the 28-Week Salmeterol Multicenter Asthma Research Trial (SMART), by Duration of Treatment:
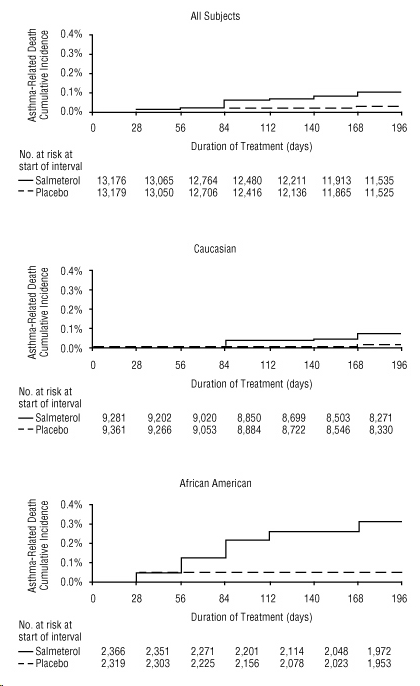
14.2 Exercise-Induced Bronchospasm
In 2 randomized, single-dose, crossover trials in adolescents and adults with EIB (N=52), 50 mcg of SEREVENT DISKUS prevented EIB when dosed 30 minutes prior to exercise. For some subjects, this protective effect against EIB was still apparent up to 8.5 hours following a single dose (Table 6).
Table 6. Results of 2 Exercise-Induced Bronchospasm Trials in Adolescents and Adults:
| SEREVENT DISKUS (N=52) | Placebo (N=52) | ||||
|---|---|---|---|---|---|
| n | % Total | n | % Total | ||
| 0.5-Hour postdose exercise challenge | % Fall in FEV1 | ||||
| <10% | 31 | 60 | 15 | 29 | |
| ≥10%, <20% | 11 | 21 | 3 | 6 | |
| ≥20% | 10 | 19 | 34 | 65 | |
| Mean maximal % fall in FEV1 (SE) | -11% (1.9) | -25% (1.8) | |||
| 8.5-Hour postdose exercise challenge | % Fall in FEV1 | ||||
| <10% | 26 | 50 | 12 | 23 | |
| ≥10%, <20% | 12 | 23 | 7 | 13 | |
| ≥20% | 14 | 27 | 33 | 63 | |
| Mean maximal % fall in FEV1 (SE) | -16% (2.0) | -27% (1.5) | |||
In 2 randomized trials in children aged 4 to 11 years with asthma and EIB (N=50), a single 50-mcg dose of SEREVENT DISKUS prevented EIB when dosed 30 minutes prior to exercise, with protection lasting up to 11.5 hours in repeat testing following this single dose in many subjects.
14.3 Chronic Obstructive Pulmonary Disease
In 2 clinical trials evaluating twice-daily treatment with SEREVENT DISKUS 50 mcg (n=336) compared with placebo (n=366) in subjects with chronic bronchitis with airflow limitation, with or without emphysema, improvements in pulmonary function endpoints were greater with salmeterol 50 mcg than with placebo. Treatment with SEREVENT DISKUS did not result in significant improvements in secondary endpoints assessing COPD symptoms in either clinical trial. Both trials were randomized, double-blind, parallel-group trials of 24 weeks' duration and were identical in design, subject entrance criteria, and overall conduct.
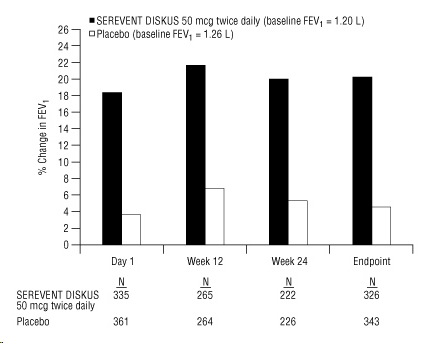
Figure 3 displays the integrated 2-hour postdose FEV1 results from the 2 clinical trials. The percent change in FEV1 refers to the change from baseline, defined as the predose value on Treatment Day 1. To account for subject withdrawals during the trial, Endpoint (last evaluable FEV1) data are provided. Subjects receiving SEREVENT DISKUS 50 mcg had significantly greater improvements in 2-hour postdose FEV1 at Endpoint (216 mL, 20%) compared with placebo (43 mL, 5%). Improvement was apparent on the first day of treatment and maintained throughout the 24 weeks of treatment.
Figure 3. Mean Percent Change from Baseline in Postdose FEV1 Integrated Data from 2 Trials of Subjects with Chronic Bronchitis and Airflow Limitation:
Onset of Action and Duration of Effect
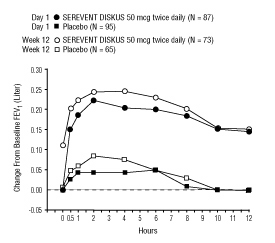
The onset of action and duration of effect of SEREVENT DISKUS were evaluated in a subset of subjects (n=87) from 1 of the 2 clinical trials discussed above. Following the first 50-mcg dose, significant improvement in pulmonary function (mean FEV1 increase of 12% or more and at least 200 mL) occurred at 2 hours. The mean time to peak bronchodilator effect was 4.75 hours. As seen in Figure 4, evidence of bronchodilatation was seen throughout the 12-hour period. Figure 4 also demonstrates that the bronchodilating effect after 12 weeks of treatment was similar to that observed after the first dose. The mean time to peak bronchodilator effect after 12 weeks of treatment was 3.27 hours.
Figure 4. Serial 12-Hour FEV1 on the First Day and at Week 12 of Treatment:
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.