SOGROYA Solution for injection Ref.[27982] Active ingredients: Somapacitan
Source: European Medicines Agency (EU) Revision Year: 2025 Publisher: Novo Nordisk A/S, Novo Allé, DK-2880 Bagsværd, Denmark
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Pituitary and hypothalamic hormones and analogues, somatropin and somatropin agonists
ATC code: H01AC07
Mechanism of action
Somapacitan is a long-acting recombinant human growth hormone derivative. It consists of 191 amino acids similar to endogenous human growth hormone, with a single substitution in the amino acid backbone (L101C) to which an albumin binding moiety has been attached. The albumin binding moiety (side-chain) consists of a fatty acid moiety and a hydrophilic spacer attached to position 101 of the protein.
The mechanism of action of somapacitan is either directly via the GH-receptor and/or indirectly via IGF-I produced in tissues throughout the body, but predominantly by the liver. When growth hormone deficiency is treated with somapacitan a normalisation of body composition (i.e., decreased body fat mass, increased lean body mass) and of metabolic action is achieved.
Somapacitan stimulates skeletal growth in paediatric patients with GHD as a result of effects on the growth plates (epiphyses) of bones, see section 5.3.
Pharmacodynamic effects
IGF-I
IGF-I is a generally accepted biomarker for efficacy in GHD.
A dose-dependent IGF-I response is induced following somapacitan administration.
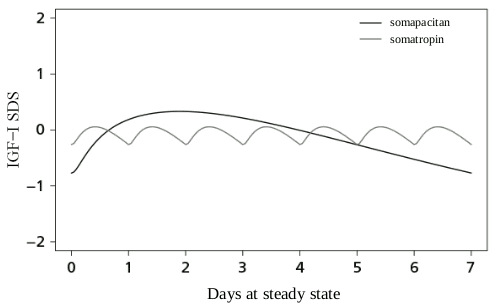
A steady state pattern in IGF-I responses is reached after 1-2 weekly doses. The IGF-I levels fluctuate during the week. The IGF-I response is maximal after 2 to 4 days. Compared with daily GH treatment, the IGF-I profile of somapacitan differs, see Figure 1.
In paediatric GHD patients somapacitan produces a dose linear IGF-I response, with a change of 0.02 mg/kg on average resulting in a change in IGF-I standard deviation score (SDS) of 0.32.
Figure 1. Model-derived IGF-I profiles during steady state of somapacitan and somatropin (based on data from adult GHD):
Clinical efficacy and safety
Paediatric GHD
REAL 4 (phase 3)
The efficacy and safety of once weekly somapacitan was evaluated in a 52 weeks randomised, multi-center, open-label, active-controlled, parallel-group phase 3 trial (REAL 4) in 200 treatment-naïve, paediatric patients with GHD. Patients were randomised to 0.16 mg/kg/week once weekly somapacitan (N=132) or 0.034 mg/kg/day once daily somatropin (N=68).
At baseline, the 200 patients had a mean age of 6.4 years (range: 2.5 to 11 years). 74.5% of the patients were male.
The annualised height velocity at week 52 was similar for somapacitan and somatropin (Table 4).
Table 4. Growth results at week 52 in paediatric patients with GHD:
| Once-weekly somapacitan (N=132) | Once-daily somatropin (N=68) | Estimate of treatment difference (95% CI) (somapacitan minus somatropin) | |
|---|---|---|---|
| Annualised height velocity (cm/year) | 11.2 | 11.7 | -0.5 [-1.1; 0.2] |
In accordance with this, changes at week 52 compared to baseline with respect to the height SDS and IGF-I SDS were also similar for somapacitan and somatropin (Table 5).
Table 5. Height SDS and IGF-I SDS in paediatric patients with GHD – 52 weeks treatment:
| Once-weekly somapacitan (N=132) | Once-daily somatropin (N=68) | Estimate of treatment difference (95% CI) (somapacitan minus somatropin) | |
|---|---|---|---|
| Height SDS, baselinea | -2.99 | -3.47 | |
| Height SDS, change from baseline | 1.25 | 1.30 | -0.05 [-0.18; 0.08] |
| IGF-I SDS, baselinea | -2.03 | -2.33 | |
| IGF-I SDS, week 52a | 0.28 | 0.10 | |
| IGF-I SDS level change from baseline | 2.36 | 2.33 | 0.03 [-0.30; 0.36] |
a Observed mean
The vast majority of paediatric patients (96.9%) in the trial achieved an average IGF-I SDS level within normal range (-2 to +2) after 52 weeks of treatment with once weekly somapacitan (Table 6). A low number of patients had average IGF-I SDS above +2 (2.3%) and no patients had average IGF-I SDS above +3.
Table 6. Average IGF-I SDS values after 52 weeks of treatment of paediatric patients with GHD with once weekly somapacitan:
| IGF-I SDS category | Week 52 average (N=132) |
|---|---|
| <-2 | 0.8% |
| -2 to 0 | 21.2% |
| 0 to +2 | 75.8% |
| +2 to +3 | 2.3% |
| >+3 | 0 |
REAL 3 (phase 2)
A total of 59 GH treatment-naïve GH-deficient paediatric patients completed a 26-week main period and a 26-week extension in a 4-arm parallel group trial with once weekly somapacitan at dose levels of 0.04, 0.08 and 0.16 mg/kg/week and active control arm of 0.034 mg/kg/day daily somatropin. The patients continued in a 104-week open-label safety extension parallel arms with somapacitan 0.16 mg/kg/week and daily somatropin 0.034 mg/kg/day. All patients were afterwards transferred to once weekly somapacitan 0.16 mg/kg/week in a 208-week long-term safety extension.
Treatment with once weekly somapcitan led to continuous treatment benefits up to at least week 208. Height SDS was -1.06 (change from baseline: 2.85) in 38 patients.
Height outcome obtained at week 208 in patients switching from 0.034 mg/kg/day daily somatropin to 0.16 mg/kg/week once weekly somapacitan at week 156 indicated that treatment benefits with daily GH treatment are maintained after switching to once weekly somapacitan.
Mean IGF-I SDS remained within the normal range for all groups.
Adult GHD
In a 34-week placebo-controlled (double-blind) and active-controlled (open) trial, 301 treatment-naïve adult patients with GHD were randomised (2:1:2) and 300 were exposed to once-weekly somapacitan or to placebo or to daily somatropin for a 34-week treatment period (main phase of the trial). The patient population had a mean age of 45.1 years (range 23-77 years; 41 patients were 65 years or above), 51.7% were females, and 69.7% had adult onset GHD.
A total of 272 adult GHD patients who completed the 34-week main phase continued in a 53-week open-label extension period. Subjects on placebo were switched to somapacitan and patients on somatropin were re-randomised (1:1) to either somapacitan or somatropin.
Observed clinical effects for the main endpoints in the main treatment phase (Table 7) and extension treatment phase (Table 8) are presented below.
Table 7. Results at 34 weeks:
| Change from baseline at 34 weeksa | somapacitan | somatropin | placebo | Difference somapacitan - placebo [95% CI] p-value | Difference somapacitan - somatropin [95% CI] |
|---|---|---|---|---|---|
| Number of subjects (Ν) | 120 | 119 | 61 | ||
| Truncal fat % (Primary endpoint) | -1.06 | -2.23 | 0.47 | -1.53 [-2.68; -0.38] 0.0090b | 1.17 [0.23; 2.11] |
| Visceral adipose tissue (cm²) | -10 | -9 | 3 | -14 [-21; -7] | -1 [-7; 4] |
| Appendicular skeletal muscle mass (g) | 558 | 462 | -121 | 679 [340; 1.019] | 96 [-182; 374] |
| Lean body mass (g) | 1.394 | 1.345 | 250 | 1144 [459; 1.829] | 49 [-513; 610] |
| IGF-I SDS level | 2.40 | 2.37 | -0.01 | 2.40 [2.09; 2.72] | 0.02 [-0.23; 0.28] |
Abbreviations: N = Number of subjects in full analysis set, CI = Confidence interval, DM=Diabetes mellitus. IGF-I SDS: Insulin-like growth factor-I standard deviation score.
a Body composition parameters are based on dual-energy X-ray absorptiometry (DXA) scanning.
b The primary analysis was a comparison of changes from baseline for somapacitan and placebo in truncal fat %.
Changes in truncal fat % from baseline to the 34 week's measurements was analysed using an analysis of covariance model with treatment, GHD onset type, sex, region, DM and sex by region by DM interaction as factors and baseline as a covariate incorporating a multiple imputation technique where missing week 34 values were imputed based on data from the placebo group.
Post-hoc subgroup analysis of changes from baseline in truncal fat percentage (%) compared to placebo at week 34 showed an estimated treatment difference (somapacitan-placebo) of -2.49% [-4.19; -0.79] in men, -0.80% [-2.99; 1.39] in women not on oral oestrogen, -1.44% [-3.97; 1.09] in women on oral oestrogen.
Table 8. Results at 87 weeks:
| Change from baseline at 87 weeksa | somapacitan/ somapacitan | somatropin/ somatropin | placebo/ somapacitan | somatropin/ somatropin | Difference somapacitan/ somapacitan vs somatropin/somatropin [95% CI] |
|---|---|---|---|---|---|
| Number of subjects (Ν) | 114 | 52 | 54 | 51 | |
| Truncal fat % | -1.51 | -2.67 | -2.28 | -1.35 | 1.15 [-0.10; 2.40] |
| Visceral adipose tissue (cm²) | -6.64 | -6.85 | -10.21 | -8.77 | 0.22 [-10; 10] |
| Appendicular skeletal muscle mass (g) | 546.11 | 449.09 | 411.05 | 575.80 | 97.02 [-362; 556] |
| Lean body mass (g) | 1,739.05 | 1,305.73 | 1,660.56 | 1,707.82 | 433.32 [-404; 1.271] |
a Body composition parameters are based on DXA scanning.
Observed and simulated IGF-I SDS levels in the clinical study
In the main phase of the clinical study IGF-I SDS values of 0 and above were overall achieved in 53% of somapacitan-treated adult GHD study patients after an 8-week dose titration period. This proportion was however lower in particular subgroups such as women on oral oestrogen (32%) and patients with childhood-onset (39%) (Table 9). Post-hoc simulation analyses indicated that the proportions of adult GHD patients achieving IGF-I SDS levels above 0 are expected to be higher in case somapacitan dose titration beyond 8 weeks would be allowed. In this simulation analysis, it was assumed that somapacitan dose titration was well-tolerated in all patients until the IGF-I SDS target range or a somapacitan dose of 8 mg per week would be achieved.
Table 9. Proportions of somapacitan-treated AGHD patients with IGF-I SDS levels above 0:
| Subgroups | Men | Women not on oral oestrogen | Women on oral oestrogen | Childhood- onset AGHD | Adult- onset AGHD | All |
|---|---|---|---|---|---|---|
| Observeda | 71% | 46% | 32% | 39% | 60% | 53% |
| Post-hoc simulations | 100% | 96% | 70% | 84% | 92% | 90% |
a The trial was designed to titrate towards a IGF-I SDS level above -0.5
Maintenance dose
Maintenance dose varies from person to person and between male and female patients. The average somapacitan maintenance dose observed in the phase 3 clinical trials was 2.4 mg/week.
Paediatric and adult GHD
Clinical safety
The safety profile of somapacitan was similar to the well-known safety profile of somatropin. No new safety issues were identified,see section 4.8.
Immunogenicity
Anti-drug antibodies (ADA) were uncommonly detected in peadiatric patients (16/132). None of these antibodies were neutralising. No evidence of ADA impact on pharmacokinetics, efficacy or safety was observed. No anti-drug antibodies were detected in adult patients.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with Sogroya in all subsets of the paediatric population in growth hormone deficiency (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Somapacitan has pharmacokinetic properties compatible with once weekly administration. The reversible binding to endogenous albumin delays elimination of somapacitan and thereby prolongs the in vivo half-life and duration of action.
The pharmacokinetics of somapacitan following subcutaneous administration have been investigated at dose levels from 0.02 to 0.16 mg/kg/week in paediatric population (2.5 to 14 years), at dose levels from 0.01 to 0.32 mg/kg in healthy adults, and in doses up to 0.12 mg/kg in patients with adult GHD.
Overall, somapacitan displays non-linear pharmacokinetics across the investigated dose range. However, in the clinically relevant dose range of somapacitan in adult GHD, somapacitan pharmacokinetics are approximately linear.
In paediatric GHD, a somapacitan dose of 0.16 mg/kg/week corresponds to an average concentration of 80.2 ng/mL and in adult GHD, somapacitan doses in the clinically relevant range correspond to average concentrations of 0.1-36.2 ng/mL.
Absorption
In adult and paediatric patients with GHD median tmax ranged from 4 to 25.5 hours at doses from 0.02 mg/kg/week to 0.16 mg/kg/week.
Steady state exposure was achieved following 1-2 weekly administration.
Absolute bioavailability of somapacitan in humans has not been investigated.
Distribution
Somapacitan is extensively bound (>99%) to plasma proteins and is expected to be distributed like albumin. Based on population PK analyses, the estimated volume of distribution (V/F) was 1.7 L in paediatric GHD patients and 14.6 L in adult GHD patients.
Elimination
Following a single dose and repeated dosing of 0.16 mg/kg/week the terminal half-life was approximately 34 hours in paediatric GHD patients.
The terminal half-life was estimated with geometric means ranging from approximately 2 to 3 days at steady state in paediatric and adult GHD patients (doses: 0.02 to 0.12 mg/kg).
Somapacitan will be present in circulation for approximately 2 weeks after the last dose. Little to no accumulation (mean accumulation ratio: 1-2) of somapacitan following multiple dosing has been observed.
Biotransformation
Somapacitan is extensively metabolised by proteolytic degradation and cleavage of the linker sequence between the peptide and albumin binder.
Somapacitan was extensively metabolised before excretion and no intact somapacitan was found neither in urine, which was the main excretion route (81%), nor in faeces where 13% of somapacitan related material was found, indicating full biotransformation before excretion.
Special populations
Paediatric GHD patients
Based on population pharmacokinetic analysis gender, race and body weight do not have a clinically meaningful effect on the pharmacokinetics following weight-based dosing.
Adult GHD patients
Age:
Subjects older than 60 years have higher exposure (29%) than younger subjects at the same somapacitan dose. A lower starting dose for subjects above 60 years is described in section 4.2.
Gender:
Female subjects and in particular female subjects on oral oestrogen, have lower exposure (53% for females on oral oestrogen and 30% for females not on oral oestrogen) than male subjects at the same somapacitan dose. A higher starting dose for females on oral oestrogen is described in section 4.2.
Race:
There was no difference in somapacitan exposure and IGF-I response between Japanese and White subjects. Despite a higher exposure in Asian Non-Japanese compared to White at the same somapacitan dose, White, Japanese and Asian Non-Japanese needed the same doses to reach similar IGF-I levels. Therefore, there is no dose adjustment recommendation based on race.
Ethnicity:
Ethnicity (Hispanic or Latino 4.5% (15 subjects received somapacitan)) was not investigated due to small sample size in the development programme.
Body weight:
Despite a higher exposure in subjects with low body weight as compared to subjects with high body weight at the same somapacitan dose, subjects needed the same doses to reach similar IGF-I levels across the body weight range 35 kg to 150 kg. Therefore, there is no dose adjustment recommendation based on body weight.
Renal impairment:
A somapacitan dose of 0.08 mg/kg at steady state resulted in higher exposures in subjects with renal impairment, most pronounced in subjects with severe renal impairment and in subjects requiring haemodialysis, where AUC0-168h ratios to normal renal function were 1.75 and 1.63, respectively. In general, somapacitan exposure tended to increase with decreasing GFR.
Higher IGF-I AUC0-168h levels were observed in subjects with moderate and severe renal impairment and subjects requiring haemodialysis, with ratios to normal renal function of 1.35, 1.40 and 1.24 respectively.
Due to the modest increase observed in IGF-I combined with the low recommended starting doses and the individual dose titration of somapacitan, there is no dose adjustment recommendation in patients with renal impairment.
Hepatic impairment:
A somapacitan dose of 0.08 mg/kg at steady state resulted in higher exposure in subjects with moderate hepatic impairment with ratios to normal hepatic function of 4.69 for AUC0-168h and 3.52 for Cmax.
Lower somapacitan stimulated IGF-I levels were observed in subjects with mild and moderate hepatic impairment compared to subjects with normal hepatic function (ratio to normal was 0.85 for mild and 0.75 for moderate).
Due to the modest decrease observed in IGF-I combined with the individual dose titration of somapacitan, there is no dose adjustment recommendation in patients with hepatic impairment.
5.3. Preclinical safety data
Preclinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeat-dose toxicity, genotoxicity or pre- and postnatal development.
No carcinogenicity studies have been performed with somapacitan.
No adverse effects were observed on male and female fertility in rats at a dose resulting in exposure at least 13 and 15-times greater than the expected maximum clinical exposure at 8 mg/week for males and females, respectively. However, irregular female oestrus cycle was seen at all doses treated.
No evidence of foetal harm was identified when pregnant rats and rabbits were administered subcutaneous somapacitan during organogenesis at doses leading to exposures well above expected exposure at the maximum clinical dose of 8 mg/week (at least 18-fold). At high doses leading to exposure at least 130-fold above the expected maximum clinical exposure at 8 mg/week, short/bent/thickened long bones were found in pups from female rats receiving somapacitan. Such findings in rats are known to resolve after birth and should be regarded as minor malformations, not permanent abnormalities.
Foetal growth was reduced when pregnant rabbits were dosed with somapacitan subcutaneously at exposures at least 9-fold above the expected exposure at the maximum clinical dose of 8 mg/week.
In lactating rats, somapacitan related material was secreted into milk but to a lower level than observed in plasma (up to 50% of level in plasma).
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.