SOLIRIS Concentrate for solution for infusion Ref.[8919] Active ingredients: Eculizumab
Source: European Medicines Agency (EU) Revision Year: 2023 Publisher: Alexion Europe SAS, 103-105 rue Anatole France, 92300 Levallois-Perret, FRANCE
Pharmacodynamic properties
Pharmacotherapeutic group: Selective immunosuppressants
ATC code: L04AJ01
Soliris is a recombinant humanised monoclonal IgG2/4k antibody that binds to the human C5 complement protein and inhibits the activation of terminal complement. The Soliris antibody contains human constant regions and murine complementarity-determining regions grafted onto the human framework light- and heavy-chain variable regions. Soliris is composed of two 448 amino acid heavy chains and two 214 amino acid light chains and has a molecular weight of approximately 148 kDa.
Soliris is produced in a murine myeloma (NS0 cell line) expression system and purified by affinity and ion exchange chromatography. The bulk drug substance manufacturing process also includes specific viral inactivation and removal steps.
Mechanism of action
Eculizumab, the active ingredient in Soliris, is a terminal complement inhibitor that specifically binds to the complement protein C5 with high affinity, thereby inhibiting its cleavage to C5a and C5b and preventing the generation of the terminal complement complex C5b-9. Eculizumab preserves the early components of complement activation that are essential for opsonization of microorganisms and clearance of immune complexes.
In PNH patients, uncontrolled terminal complement activation and the resulting complement-mediated intravascular haemolysis are blocked with Soliris treatment. In most PNH patients, eculizumab serum concentrations of approximately 35 microgram/mL are sufficient for essentially complete inhibition of terminal complement-mediated intravascular haemolysis. In PNH, chronic administration of Soliris resulted in a rapid and sustained reduction in complement-mediated haemolytic activity.
In aHUS patients, uncontrolled terminal complement activation and the resulting complement-mediated thrombotic microangiopathy are blocked with Soliris treatment. All patients treated with Soliris when administered as recommended demonstrated rapid and sustained reduction in terminal complement activity. In all aHUS patients, eculizumab serum concentrations of approximately 50-100 microgram/mL are sufficient for essentially complete inhibition of terminal complement activity.
In aHUS, chronic administration of Soliris resulted in a rapid and sustained reduction in complement-mediated thrombotic microangiopathy.
In refractory gMG patients, uncontrolled terminal complement activation causes membrane attack complex (MAC) dependent lysis and C5a-dependent inflammation at the Neuromuscular Junction (NMJ) leading to failure of neuromuscular transmission. Chronic administration of Soliris results in immediate, complete, and sustained inhibition of terminal complement activity (eculizumab serum concentrations ≥116 microgram/ml).
In patients with NMOSD, uncontrolled terminal complement activation caused by autoantibodies against AQP4 leads to the formation of the MAC and C5a-dependent inflammation which results in astrocyte necrosis and increased permeability of the blood brain barrier, as well as death of the surrounding oligodendrocytes and neurons. Chronic administration of Soliris results in immediate, complete, and sustained inhibition of terminal complement activity (eculizumab serum concentrations ≥116 microgram/ml).
Clinical efficacy and safety
Paroxysmal Nocturnal Haemoglobinuria
The safety and efficacy of Soliris in PNH patients with haemolysis were assessed in a randomized, double-blind, placebo-controlled 26 week study (C04-001). PNH patients were also treated with Soliris in a single arm 52 week study (C04-002), and in a long term extension study (E05-001). Patients received meningococcal vaccination prior to receipt of Soliris. In all studies, the dose of eculizumab was 600 mg every 7 ± 2 days for 4 weeks, followed by 900 mg 7 ± 2 days later, then 900 mg every 14 ± 2 days for the study duration. Soliris was administered as an intravenous infusion over 25–45 minutes (35 minutes ± 10 minutes). An observational non-interventional Registry in patients with PNH (M07-001) was also initiated to characterize the natural history of PNH in untreated patients and the clinical outcomes during Soliris treatment.
In study C04-001 (TRIUMPH) PNH patients with at least 4 transfusions in the prior 12 months, flow cytometric confirmation of at least 10% PNH cells and platelet counts of at least 100,000/microliter were randomized to either Soliris (n=43) or placebo (n=44). Prior to randomization, all patients underwent an initial observation period to confirm the need for RBC transfusion and to identify the haemoglobin concentration (the "set-point") which would define each patient's haemoglobin stabilization and transfusion outcomes. The haemoglobin set-point was less than or equal to 9 g/dL in patients with symptoms and was less than or equal to 7 g/dL in patients without symptoms. Primary efficacy endpoints were haemoglobin stabilization (patients who maintained a haemoglobin concentration above the haemoglobin set-point and avoid any RBC transfusion for the entire 26 week period) and blood transfusion requirement. Fatigue and health-related quality of life were relevant secondary endpoints. Haemolysis was monitored mainly by the measurement of serum LDH levels, and the proportion of PNH RBCs was monitored by flow cytometry. Patients receiving anticoagulants and systemic corticosteroids at baseline continued these medications. Major baseline characteristics were balanced (see Table 2).
In the non-controlled study C04-002 (SHEPHERD), PNH patients with at least one transfusion in the prior 24 months and at least 30,000 platelets/microliter received Soliris over a 52-week period. Concomitant medications included anti-thrombotic agents in 63% of the patients and systemic corticosteroids in 40% of the patients. Baseline characteristics are shown in Table 2.
Table 2. Patient Demographics and Characteristics in C04-001 and C04-002:
| C04-001 | C04-002 | ||
|---|---|---|---|
| Parameter | Placebo N=44 | Soliris N=43 | Soliris N=97 |
| Mean Age (SD) | 38,4 (13,4) | 42,1 (15,5) | 41,1 (14,4) |
| Gender - Female (%) | 29 (65,9) | 23 (53,5) | 49 (50,5) |
| History of Aplastic Anaemia or MDS (%) | 12 (27,3) | 8 (18,7) | 29 (29,9) |
| Concomitant Anticoagulants (%) | 20 (45,5) | 24 (55,8) | 59 (61) |
| Concomitant Steroids/Immunosuppressant Treatments (%) | 16 (36,4) | 14 (32,6) | 46 (47,4) |
| Discontinued treatment | 10 | 2 | 1 |
| PRBC in previous 12 months (median (Q1,Q3)) | 17,0 (13,5, 25,0) | 18,0 (12,0, 24,0) | 8,0 (4,0, 24,0)4 |
| Mean Hgb level (g/dL) at setpoint (SD) | 7,7 (0,75) | 7,8 (0,79) | N/A |
| Pre-treatment LDH levels (median, U/L) | 2.234,5 | 2.032,0 | 2.051,0 |
| Free Haemoglobin at baseline (median, mg/dL) | 46,2 | 40,5 | 34,9 |
In TRIUMPH, study patients treated with Soliris had significantly reduced (p<0.001) haemolysis resulting in improvements in anaemia as indicated by increased haemoglobin stabilization and reduced need for RBC transfusions compared to placebo treated patients (see Table 3). These effects were seen among patients within each of the three pre-study RBC transfusion strata (4-14 units; 15-25 units; >25 units). After 3 weeks of Soliris treatment, patients reported less fatigue and improved health-related quality of life. Because of the study sample size and duration, the effects of Soliris on thrombotic events could not be determined. In SHEPHERD study, 96 of the 97 enrolled patients completed the study (one patient died following a thrombotic event). A reduction in intravascular haemolysis as measured by serum LDH levels was sustained for the treatment period and resulted in increased transfusion avoidance, a reduced need for RBC transfusion and less fatigue. See Table 3.
Table 3. Efficacy Outcomes in C04-001 and C04-002:
| C04-001 | C04-002* | ||||
|---|---|---|---|---|---|
| Placebo N=44 | Soliris N=43 | P–Value | Soliris N=97 | P–Value | |
| Percentage of patients with stabilized Haemoglobin levels at end of study | 0 | 49 | <0,001 | N/A | |
| PRBC transfused during treatment (median) | 10 | 0 | <0,001 | 0 | <0,001 |
| Transfusion Avoidance during treatment (%) | 0 | 51 | <0,001 | 51 | <0,001 |
| LDH levels at end of study (median, U/L) | 2.167 | 239 | <0,001 | 269 | <0,001 |
| LDH AUC at end of study (median, U/L x Day) | 411.822 | 58.587 | <0,001 | -632.264 | <0,001 |
| Free Haemoglobin at end of study (median, mg/dL) | 62 | 5 | <0,001 | 5 | <0,001 |
| FACIT-Fatigue (effect size) | 1,12 | <0,001 | 1,14 | <0,001 | |
* Results from study C04-002 refer to pre-versus post-treatment comparisons.
From the 195 patients that originated in C04-001, C04-002 and other initial studies, Soliris-treated PNH patients were enrolled in a long term extension study (E05-001). All patients sustained a reduction in intravascular haemolysis over a total Soliris exposure time ranging from 10 to 54 months. There were fewer thrombotic events with Soliris treatment than during the same period of time prior to treatment. However, this finding was shown in non-controlled clinical trials.
The PNH registry (M07-001) was used to evaluate the efficacy of Soliris in PNH patients with no history of RBC transfusion. These patients had high disease activity as defined by elevated haemolysis (LDH ≥1.5x ULN) and the presence of related clinical symptom(s): fatigue, haemoglobinuria, abdominal pain, shortness of breath (dyspnoea), anaemia (haemoglobin <100 g/L), major adverse vascular event (including thrombosis), dysphagia, or erectile dysfunction.
In the PNH Registry, patients treated with Soliris were observed to have a reduction in haemolysis and associated symptoms. At 6 months, patients treated with Soliris with no history of RBC transfusion had significantly (p<0.001) reduced LDH levels (median LDH of 305 U/L; Table 4). Furthermore, 74% of the patients without a history of transfusion and treated with Soliris experienced clinically meaningful improvements in FACIT-Fatigue score (i.e. increase by 4 points or more) and 84% in EORTC fatigue score (i.e. decrease by 10 points or more).
Table 4. Efficacy Outcomes (LDH level and FACIT-Fatigue) in Patients with PNH with No History of Transfusion in M07-001:
| M07-001 | |
|---|---|
| Parameter | Soliris No transfusion |
| LDH level at baseline (median, U/L) | N=43 1447 |
| LDH level at 6 months (median, U/L) | N=36 305 |
| FACIT-Fatigue score at baseline (median) | N=25 32 |
| FACIT-Fatigue score at last available assessment (median) | N=31 44 |
FACIT-Fatigue is measured on a scale of 0-52, with higher values indicating less fatigue
Atypical Haemolytic Uremic Syndrome
Data from 100 patients in four prospective controlled studies, three in adult and adolescent patients (C08-002A/B C08-003A/B, C10-004) one in paediatric and adolescent patients (C10-003) and 30 patients in one retrospective study (C09-001r) were used to evaluate the efficacy of Soliris in the treatment of aHUS.
Study C08-002A/B was a prospective, controlled, open-label study which accrued patients in the early phase of aHUS with evidence of clinical thrombotic microangiopathy manifestations with platelet count ≤150 x 109/L despite PE/PI, and LDH and serum creatinine above upper limits of normal. Study C08-003A/B was a prospective, controlled, open-label study which accrued patients with longer term aHUS without apparent evidence of clinical thrombotic microangiopathy manifestations and receiving chronic PE/PI (≥1 PE/PI treatment every two weeks and no more than 3 PE/PI treatments/week for at least 8 weeks before the first dose). Patients in both prospective studies were treated with Soliris for 26 weeks and most patients enrolled into a long-term, open-label extension study. All patients enrolled in both prospective studies had an ADAMTS-13 level above 5%.
Patients received meningococcal vaccination prior to receipt of Soliris or received prophylactic treatment with appropriate antibiotics until 2 weeks after vaccination. In all studies, the dose of Soliris in adult and adolescent aHUS patients was 900 mg every 7 ± 2 days for 4 weeks, followed by 1,200 mg 7 ± 2 days later, then 1,200 mg every 14 ± 2 days for the study duration. Soliris was administered as an intravenous infusion over 35 minutes. The dosing regimen in paediatric patients and adolescents weighing less than 40 kg was defined based on a pharmacokinetic (PK) simulation that identified the recommended dose and schedule based on body weight (see section 4.2).
Primary endpoints included platelet count change from baseline in study C08-002A/B and thrombotic microangiopathy (TMA) event-free status in study C08-003A/B. Additional endpoints included TMA intervention rate, haematologic normalization, complete TMA response, changes in LDH, renal function and quality of life. TMA-event free status was defined as the absence for at least 12 weeks of the following: decrease in platelet count of >25% from baseline, PE/PI, and new dialysis. TMA interventions were defined as PE/PI or new dialysis. Haematologic normalization was defined as normalization of platelet counts and LDH levels sustained for ≥2 consecutive measurements for ≥4 weeks. Complete TMA response was defined as haematologic normalization and a ≥25% reduction in serum creatinine sustained in ≥2 consecutive measurements for ≥4 weeks. Baseline characteristics are shown in Table 5.
Table 5. Patient Demographics and Characteristics in C08-002A/B and C08-003A/B:
| Parameter | C08-002A/B | C08-003A/B |
|---|---|---|
| Soliris N=17 | Soliris N=20 | |
| Time from first diagnosis until screening in months, median (min, max) | 10 (0,26, 236) | 48 (0,66, 286) |
| Time from current clinical TMA manifestation until screening in months, median (min, max) | <1 (<1, 4) | 9 (1, 45) |
| Number of PE/PI sessions for current clinical TMA manifestation, median (min, max) | 17 (2, 37) | 62 (20, 230) |
| Number of PE/PI sessions in 7 days prior to first dose of eculizumab, median (min, max) | 6 (0, 7) | 2 (1, 3) |
| Baseline platelet coun (×109/L), mean (SD) | 109 (32) | 228 (78) |
| Baseline LDH (U/L), mean (SD) | 323 (138) | 223 (70) |
| Patients without identified mutation, n (%) | 4 (24) | 6 (30) |
Patients in aHUS Study C08-002 A/B received Soliris for a minimum of 26 weeks. After completion of the initial 26-week treatment period, most patients continued to receive Soliris by enrolling into an extension study. In aHUS Study C08-002A/B, the median duration of Soliris therapy was approximately100 weeks (range: 2 weeks to 145 weeks).
A reduction in terminal complement activity and an increase in platelet count relative to baseline were observed after commencement of Soliris. Reduction in terminal complement activity was observed in all patients after commencement of Soliris. Table 6 summarizes the efficacy results for aHUS Study C08- 002A/B. All rates of efficacy endpoints improved or were maintained through 2 years of treatment. Complete TMA response was maintained by all responders. When treatment was continued for more than 26 weeks, two additional patients achieved and maintained Complete TMA response due to normalization of LDH (1 patient) and a decrease in serum creatinine (2 patients).
Renal function, as measured by eGFR, was improved and maintained during Soliris therapy. Four of the five patients who required dialysis at study entry were able to discontinue dialysis for the duration of Soliris treatment, and one patient developed a new dialysis requirement. Patients reported improved health-related quality of life (QoL).
In aHUS Study C08-002A/B, responses to Soliris were similar in patients with and without identified mutations in genes encoding complement regulatory factor proteins.
Patients in aHUS study C08-003A/B received Soliris for a minimum of 26 weeks. After completion of the initial 26-week treatment period, most patients continued to receive Soliris by enrolling into an extension study. In aHUS Study C08-003A/B, the median duration of Soliris therapy was approximately 114 weeks (range: 26 to 129 weeks). Table 6 summarizes the efficacy results for aHUS Study C08-003A/B. In aHUS Study C08-003A/B, responses to Soliris were similar in patients with and without identified mutations in genes encoding complement regulatory factor proteins. Reduction in terminal complement activity was observed in all patients after commencement of Soliris. All rates of efficacy endpoints improved or were maintained through 2 years of treatment. Complete TMA response was maintained by all responders. When treatment was continued for more than 26 weeks, six additional patients achieved and maintained Complete TMA response due to a decrease in serum creatinine. No patient required new dialysis with Soliris. Renal function, as measured by median eGFR, increased during Soliris therapy.
Table 6. Efficacy Outcomes in Prospective aHUS Studies C08-002A/B and C08-003A/B:
| C08-002A/B N=17 | C08-003A/B N=20 | |||
|---|---|---|---|---|
| At 26 weeks | At 2 years1 | At 26 weeks | At 2 years1 | |
| Normalization of platelet count | 14 (82) | 15 (88) | 18 (90) | 18 (90) |
| All patients, n (%) (95% CI) | (57-96) | (64-99) | (68-99) | (68-99) |
| Patients with abnormal baseline, n/n (%) | 13/15 (87) | 13/15 (87) | 1/3 (33) | 1/3 (33) |
| TMA event-free status, n (%) (95% CI) | 15 (88) (64-99) | 15 (88) (64-99) | 16 (80) (56-94) | 19 (95) (75-99) |
| TMA intervention rate | ||||
| Daily pre- eculizumab rate, median (min, max) | 0,88 (0,04, 1,59) | 0,88 (0,04, 1,59) | 0,23 (0,05, 1,09) | 0,23 (0,05, 1,09) |
| Daily during- eculizumab rate, median (min, max) | 0 (0, 0,31) | 0 (0, 0,31) | 0 | 0 |
| P-value | P<0,0001 | P<0,0001 | P <0,0001 | P<0,0001 |
| CKD improvement by ≥1 stage, n (%) (95% CI) | 10 (59) (33-82) | 12 (71) (44-90) | 7 (35) (15-59) | 12 (60) (36-81) |
| eGFR change mL/min/1,73 m²: median (range) | 20 (-1, 98) | 28 (3, 82) | 5 (-1, 20) | 11 (-42, 30) |
| eGFR improvement ≥15 mL/min/1,73 m², n (%) (95% CI) | 8 (47) (23-72) | 10 (59) (33-82) | 1 (5) (0-25) | 8 (40) (19-64) |
| Change in Hgb >20 g/L, n (%) (95% CI) | 11 (65) (38-86)2 | 13 (76) (50-93) | 9 (45) (23-68)3 | 13 (65) (41-85) |
| Haematologic normalization, n (%) (95% CI) | 13 (76) (50-93) | 15 (88) (64-99) | 18 (90) (68-99) | 18 (90) (68-99) |
| Complete TMA response, n (%) (95% CI) | 11 (65) (38-86) | 13 (76) (50-93) | 5 (25) (9-49) | 11 (55) (32-77) |
1 At data cut off (20 April 2012)
2 Study C08-002: 3 patients received ESA which was discontinued after eculizumab initiation
3 Study C08-003: 8 patients received ESA which was discontinued in 3 of them during eculizumab therapy
aHUS Study C10-004 enrolled 41 patients who displayed signs of thrombotic microangiopathy (TMA). In order to qualify for enrolment, patients were required to have a platelet count < lower limit of normal range (LLN), evidence of haemolysis such as an elevation in serum LDH, and serum creatinine above the upper limits of normal, without the need for chronic dialysis. The median patient age was 35 (range: 18 to 80 years). All patients enrolled in aHUS Study C10-004 had an ADAMTS-13 level above 5%. Fifty-one percent of patients had an identified complement regulatory factor mutation or auto-antibody. A total of 35 patients received PE/PI prior to eculizumab. Table 7 summarizes the key baseline clinical and disease-related characteristics of patients enrolled in aHUS C10-004.
Table 7. Baseline Characteristics of Patients Enrolled in aHUS Study C10-004:
| Parameter | aHUS Study C10-004 N=41 |
|---|---|
| Time from aHUS diagnosis to first study dose (months), median (min, max) | 0,79 (0,03, 311) |
| Time from current clinical TMA manifestation until first study dose (months), median ( min, max) | 0,52 (0,03,19) |
| Baseline platelet count (×109/L), median (min, max) | 125 (16, 332) |
| Baseline LDH (U/L), median (min, max) | 375 (131, 3318) |
| Baseline eGFR (mL/min/1,73 m²), median (min, max) | 10 (6, 53) |
Patients in aHUS Study C10-004 received Soliris for a minimum of 26 weeks. After completion of the initial 26-week treatment period, most patients elected to continue on chronic dosing.
Reduction in terminal complement activity and an increase in platelet count relative to baseline were observed after commencement of Soliris. Soliris reduced signs of complement-mediated TMA activity, as shown by an increase in mean platelet counts from baseline to 26 weeks. In aHUS C10-004, mean (±SD) platelet count increased from 119 ± 66 x109/L at baseline to 200 ± 84 x109/L by one week; this effect was maintained through 26 weeks (mean platelet count (±SD) at week 26: 252 ± 70 x109/L). Renal function, as measured by eGFR, was improved during Soliris therapy. Twenty of the 24 patients who required dialysis at baseline were able to discontinue dialysis during Soliris treatment. Table 8 summarizes the efficacy results for aHUS study C10-004.
Table 8. Efficacy Outcomes in Prospective aHUS Study C10-004:
| Efficacy Parameter | aHUS Study C10-004 (N=41) At 26-weeks |
|---|---|
| Change in platelet count through week 26 (109/L) | 111 (-122, 362) |
| Hematologic Normalization, n (%) | 36 (88) |
| Median duration of hematologic normalization, weeks (range)1 | 46 (10, 74) |
| Complete TMA response, n (%) | 23 (56) |
| Median duration of complete TMA response, weeks (range)1 | 42 (6, 74) |
| TMA Event-free Status, n (%) | 37 (90) |
| 95% CI | 77, 97 |
| Daily TMA Intervention Rate, median (range) | |
| Before eculizumab | 0,63 (0, 1,38) |
| On eculizumab treatment | 0 (0, 0,58) |
1 Through data cut-off (September 4, 2012), with median duration of Soliris therapy of 50 weeks (range: 13 weeks to 86 weeks).
Longer term treatment with Soliris (median 52 weeks ranging from 15 to 126 weeks) was associated with an increased rate of clinically meaningful improvements in adult patients with aHUS. When Soliris treatment was continued for more than 26 weeks, three additional patients (63% of patients in total) achieved Complete TMA response and four additional patients (98% of patients in total) achieved hematologic normalization. At the last evaluation, 25 of 41 patients (61%) achieved eGFR improvement of ≥15 mL/min/1.73 m² from baseline.
Refractory Generalized Myasthenia Gravis
Data from 139 patients in two prospective controlled studies (Studies C08-001 and ECU-MG-301), and one open-label extension trial (Study ECU-MG-302) were used to evaluate the efficacy of Soliris in the treatment of patients with refractory gMG.
Study ECU-MG-301 (REGAIN) was a 26-week double-blind, randomized, placebo-controlled, multi- center Phase 3 study of Soliris in patients who had failed previous therapies and remain symptomatic. One hundred and eighteen (118) of the 125 (94%) patients completed the 26-week treatment period and 117 (94%) patients subsequently enrolled in Study ECU-MG-302, an open-label, multi-center long-term safety and efficacy extension study in which all patients received Soliris treatment.
In Study ECU-MG-301, gMG patients with a positive serologic test for anti-AChR antibodies, MGFA (Myasthenia Gravis Foundation of America) clinical classification class II to IV and MG-ADL total score ≥6 were randomized to either Soliris (n=62) or placebo (n=63). All patients included in the trial were refractory gMG patients and met the following predefined criteria:
1) Failed treatment for at least one year with 2 or more immunosuppressant therapies (either in combination or as monotherapy), ie, patients continued to have impairment in activities of daily living despite immunosuppressant therapies
OR
2) Failed at least one immunosuppressant therapy and required chronic plasma exchange or IVIg to control symptoms, ie, patients require PE or IVIg on a regular basis for the management of muscle weakness at least every 3 months over previous 12 months.
Patients received meningococcal vaccination prior to initiating treatment with Soliris or received prophylactic treatment with appropriate antibiotics until 2 weeks after vaccination. In Studies ECU-MG-301 and ECU-MG-302, the dose of Soliris in adult refractory gMG patients was 900 mg every 7 ± 2 days for 4 weeks, followed by 1200 mg at Week 5 ± 2 days, then 1,200 mg every 14 ± 2 days for the study duration. Soliris was administered as an intravenous infusion over 35 minutes.
Table 9 presents the baseline characteristics of the refractory gMG patients enrolled in Study ECU-MG-301.
Table 9. Patient Demographic and Characteristics in Study ECU-MG-301:
| Soliris (n=62) | Placebo (n=63) | |
|---|---|---|
| Age at MG Diagnosis (years), Mean (min, max) | 38,0 (5,9, 70,8) | 38,1 (7,7, 78,0) |
| Female, n (%) | 41 (66,1) | 41 (65,1) |
| Duration of MG (years), Mean (min, max) | 9,9 (1,3, 29,7) | 9,2 (1,0, 33,8) |
| Baseline MG-ADL Score | ||
| Mean (SD) | 10,5 (3,06) | 9,9 (2,58) |
| Median | 10,0 | 9,0 |
| Baseline QMG Score | ||
| Mean (SD) | 17,3 (5,10) | 16,9 (5,56) |
| Median | 17,0 | 16,0 |
| ≥3 Prior Immunosuppressive Therapies* since diagnosis, n (%) | 31 (50,0) | 34 (54,0) |
| Number of patients with prior exacerbations since diagnosis, n (%) | 46 (74,2) | 52 (82,5) |
| Number of patients with prior MG crisis since diagnosis, n (%) | 13 (21,0) | 10 (15,9) |
| Any prior ventilator support since diagnosis, n (%) | 15 (24,2) | 14 (22,2) |
| Any prior intubation since diagnosis (MGFA class V), n (%) | 11 (17,7) | 9 (14,3) |
* Immunosuppressant's included, but are not limited to, corticosteroids, azathioprine, mycophenolate, methotrexate, cyclosporine, tacrolimus, or cyclophosphamide.
The primary endpoint for Study ECU-MG-301 was the change from baseline in the MG Activities of Daily Living Profile (MG-ADL – a patient reported outcome measure validated in gMG) total score at Week 26. The primary analysis of the MG-ADL was a Worst-Rank ANCOVA with a mean rank of 56.6 for Soliris and 68.3 for placebo, based on 125 study patients (p=0.0698).
The key secondary endpoint was the change from baseline in the Quantitative MG Scoring System (QMG – a physician reported outcome measure validated in gMG) total score at Week 26. The primary analysis of the QMG was a Worst-Rank ANCOVA with a mean rank of 54.7 for Soliris and 70.7 for placebo, based on 125 study patients (p=0.0129).
Efficacy outcomes for the pre-specified repeated measures analyses of the primary and secondary endpoints are provided in Table 10.
Table 10. ECU-MG-301 Efficacy Outcomes Change from Baseline to Week 26:
| Efficacy Endpoints: Total score change from baseline at Week 26 | Soliris (n=62) (SEM) | Placebo (n=63) (SEM) | Soliris change relative to placebo – LS Mean Difference (95% CI) | p-value (using repeated measures analysis) |
|---|---|---|---|---|
| MG-ADL | -4,2 (0,49) | -2,3 (0,48) | -1,9 (-3,3, -0,6) | 0,0058 |
| QMG | -4,6 (0,60) | -1,6 (0,59) | -3,0 (-4,6, -1,3) | 0,0006 |
| MGC | -8,1 (0,96) | -4,8 (0,94) | -3,4 (-6,0, -0,7) | 0,0134 |
| MG-QoL-15 | -12,6 (1,52) | -5,4 (1,49) | -7,2 (-11,5, -3,0) | 0,0010 |
SEM = Standard Error of the Mean CI = Confidence Interval, MGC = Myasthenia Gravis Composite, MG-QoL15 = Myasthenia Gravis Qualtiy of Life 15
In Study ECU-MG-301, a clinical responder in the MG-ADL total score was defined as having at least a 3-point improvement. The proportion of clinical responders at Week 26 with no rescue therapy was 59.7% on Soliris compared with 39.7% on placebo (p=0.0229).
In Study ECU-MG-301, a clinical responder in the QMG total score was defined as having at least a 5-point improvement. The proportion of clinical responders at Week 26 with no rescue therapy was 45.2% on Soliris compared with 19% on placebo (p=0.0018).
Table 11 presents an overview of the patients reporting clinical deterioration and patients requiring rescue therapy over the 26 weeks.
Table 11. Clinical deterioration and rescue therapy in ECU-MG-301:
| Variable | Statistic | Placebo (N=63) | Soliris (N=62) |
|---|---|---|---|
| Total number of patients reporting clinical deterioration | n (%) | 15 (23,8) | 6 (9,7) |
| Total number of patients requiring rescue therapy | n (%) | 12 (19,0) | 6 (9,7) |
Of the 125 patients enrolled in ECU-MG-301, 117 patients subsequently enrolled in a long-term extension study (Study ECU-MG-302), in which all received Soliris. Patients that were previously treated with Soliris in Study ECU-MG-301 continued to demonstrate a sustained effect of Soliris on all measures (MG-ADL, QMG, MGC and MG-QoL15) over an additional 130 weeks of treatment with eculizumab in Study ECU-MG-302. For patients who received placebo in Study ECU-MG-301 (placebo/eculizumab arm of Study ECU-MG-302), improvement occurred after initiating treatment with eculizumab and was maintained for more than 130 weeks in Study ECU-MG-302. Figure 1 presents the change from baseline in both MG-ADL (A) and QMG (B) after 26 weeks of treatment in Study ECU-MG-301 and after 130 weeks of treatment (n=80 patients) in Study ECU-MG-302.
Figure 1. Mean changes from baseline in MG-ADL (1A) and QMG (1B) over Studies ECU-MG-301 and ECU-MG-302:
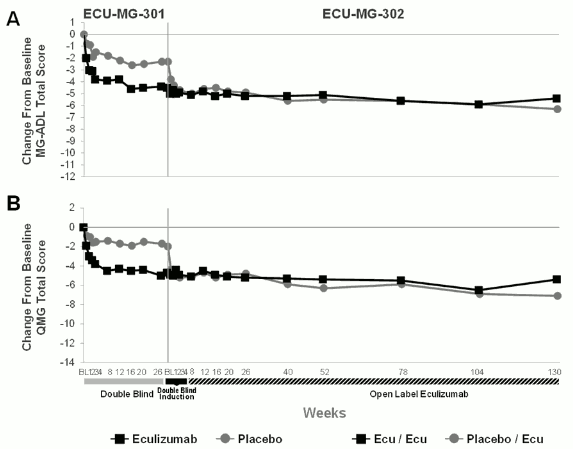
In Study ECU-MG-302, physicians had the option to adjust background immunosuppressant therapies. In this setting, 65.0% of patients decreased their daily dose of at least 1 immunosuppressive therapy (IST); 43.6% of patients stopped an existing IST. The most common reason for change in IST therapy was improvement in MG symptoms.
Twenty-two (22) (17.6%) elderly refractory gMG patients (>65 years of age) were treated with Soliris in the clinical trials. No substantial differences were seen in safety and efficacy related to age.
Neuromyelitis Optica Spectrum Disorder
Data from 143 patients in one controlled study (Study ECU-NMO-301) and from 119 patients who continued in one open-label extension trial (Study ECU-NMO-302) were used to evaluate the efficacy and safety of Soliris in the treatment of patients with NMOSD.
Study ECU-NMO-301 was a double-blind, randomized, placebo-controlled, multi-center, Phase 3 study of Soliris in patients with NMOSD.
In Study ECU-NMO-301, patients with NMOSD with a positive serologic test for anti-AQP4 antibodies, a history of at least 2 relapses in last 12 months or 3 relapses in the last 24 months with at least 1 relapse in the 12 months prior to screening and an Expanded Disability Status Scale (EDSS) score ≤7, were randomized 2:1 to either Soliris (n=96) or placebo (n=47). Patients were permitted to receive background immunosuppressant therapies at stable dose during the study, with the exclusion of rituximab and mitoxantrone.
Patients either received meningococcal vaccination at least 2 weeks prior to initiating treatment with Soliris or received prophylactic treatment with appropriate antibiotics until 2 weeks after vaccination. In the eculizumab NMOSD clinical development program, the dose of Soliris in adult patients with NMOSD was 900 mg every 7 ± 2 days for 4 weeks, followed by 1200 mg at Week 5 ± 2 days, then 1200 mg every 14 ± 2 days for the study duration. Soliris was administered as an intravenous infusion over 35 minutes.
The majority (90.9%) of patients were female. Approximately half were White (49.0%). The median age at first dose of study drug was 45 years.
Table 12. Patient Disease History and Baseline Characteristics in Study ECU-NMO-301:
| Variable | Statistic | Placebo (N=47) | Eculizumab (N=96) | Total (N=143) |
|---|---|---|---|---|
| NMOSD History | ||||
| Age at NMOSD Initial Clinical Presentation (years) | Mean (SD) | 38,5 (14,98) | 35,8 (14,03) | 36,6 (14,35) |
| Median | 38,0 | 35,5 | 36,0 | |
| Min, Max | 12, 73 | 5, 66 | 5, 73 | |
| Time from NMOSD initial clinical presentation to first dose of study drug (years) | Mean (SD) | 6,601 (6,5863) | 8,156 (8,5792) | 7,645 (7,9894) |
| Median | 3,760 | 5,030 | 4,800 | |
| Min, Max | 0,51, 29,10 | 0,41, 44,85 | 0,41, 44,85 | |
| Historical Annualized Relapse Rate within 24 months prior to Screening | Mean (SD) | 2,07 (1,037) | 1,94 (0,896) | 1,99 (0,943) |
| Median | 1,92 | 1,85 | 1,92 | |
| Min, Max | 1,0, 6,4 | 1,0, 5,7 | 1,0, 6,4 | |
| Baseline characteristics | ||||
| Baseline EDSS score | Mean (SD) | 4,26 (1,510) | 4,15 (1,646) | 4,18 (1,598) |
| Median | 4,00 | 4,00 | 4,00 | |
| Min, Max | 1,0, 6,5 | 1,0, 7,0 | 1,0, 7,0 | |
| No IST usage at baseline | n (%) | 13 (27,7) | 21 (21,9) | 34 (23,8) |
Abbreviations: ARR = adjudicated relapse rate; EDSS = Expanded Disability Status Scale; IST = immunosupressant therapy; Max = maximum; Min = minimum; NMOSD = neuromyelitis optica spectrum disorder; SD = standard deviation.
The primary endpoint for Study ECU-NMO-301 was the time to first on-trial relapse as adjudicated by an independent committee who were blinded to treatment. A significant effect on the time to first adjudicated On-trial Relapse was observed for eculizumab compared with placebo (relative risk reduction 94%; hazard ratio 0.058; p<0.0001) (Figure 2). Soliris-treated patients experienced similar improvement in time to first adjudicated on-trial relapse with or without concomitant IST treatment.
Figure 2. Kaplan-Meier Survival Estimates for Time to First Adjudicated On-Trial Relapse in Study ECU-NMO-301 – Full Analysis Set:
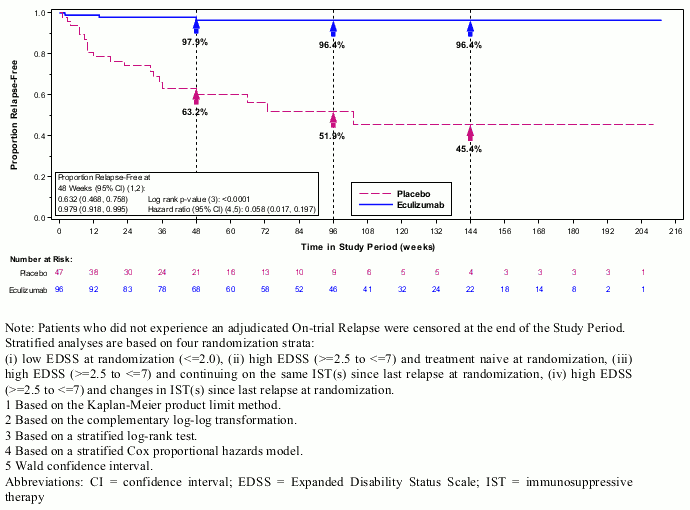
Note: Patients who did not experience an adjudicated On-trial Relapse were censored at the end of the Study Period.
Stratified analyses are based on four randomization strata:
(i) low EDSS at randomization (<=2.0), (ii) high EDSS (>=2.5 to ≤7) and treatment naive at randomization, (iii) high EDSS (>=2.5 to ≤7) and continuing on the same IST(s) since last relapse at randomization, (iv) high EDSS (>=2.5 to ≤7) and changes in IST(s) since last relapse at randomization.
1 Based on the Kaplan-Meier product limit method.
2 Based on the complementary log-log transformation.
3 Based on a stratified log-rank test.
4 Based on a stratified Cox proportional hazards model.
5 Wald confidence interval.
Abbreviations: CI = confidence interval; EDSS = Expanded Disability Status Scale; IST = immunosuppressive therapy
The adjudicated on-trial annualized relapse rate (ARR) ratio (95% CI) for eculizumab compared with placebo was 0.045 (0.013, 0.151), representing a 95.5% relative reduction in adjudicated On-trial ARR for patients treated with eculizumab compared with placebo (p<0.0001) (Table 13).
Table 13. Adjudicated On-trial Annualized Relapse Rate in Study ECU-NMO-301 – Full Analysis Set:
| Variable | Statistic | Placebo (N=47) | Eculizumab (N=96) |
|---|---|---|---|
| Total number of relapses | Sum | 21 | 3 |
| Total number of patient-years in study period | n | 52,41 | 171,32 |
| Adjusted adjudicated ARRa | Rate | 0,350 | 0,016 |
| 95% CI | 0,199, 0,616 | 0,005, 0,050 | |
| Treatment effecta | Rate ratio (eculizumab/placebo) | - | 0,045 |
| 95% CI | - | 0,013, 0,151 | |
| p-value | - | <0,0001 |
a Based on a Poisson regression adjusted for randomization strata and historical ARR in 24 months prior to Screening.
Abbreviations: ARR = annualized relapse rate; CI = confidence interval.
Compared to placebo-treated patients, Soliris-treated patients had reduced annualized rates of hospitalizations (0.04 for Soliris versus 0.31 for placebo), of intravenous corticosteroid administrations to treat acute relapses (0.07 for Soliris versus 0.42 for placebo), and of plasma exchange treatments (0.02 for Soliris versus 0.19 for placebo).
The distribution of changes from Baseline to End of Study on other secondary endpoints favored eculizumab treatment over placebo across all neurologic disability (EDSS score [p=0.0597] and mRS [nominal p=0.0154]), functional disability (HAI [nominal p=0.0002]) and quality of life (EQ-5D VAS [nominal p=0.0309] and EQ-5D Index [nominal p= 0.0077]) measures.
An interim analysis of Study ECU-NMO-302 demonstrates a significant and clinically meaningful reduction in On-trial ARR (as determined by the Treating Physician) on eculizumab treatment, based on the median (min, max) change (-1.829 [-6.38, 1.63], p<0.0001) from historical ARR (24 months prior to screening in Study ECU-NMO-301).
In Study ECU-NMO-302, physicians had the option to adjust background immunosuppressant therapies. In this setting, the most common change in immunosuppressant therapy was decreased immunosuppressant therapy dose, which occurred in 18.5% of patients. Further, 6.7% of patients stopped an existing IST.
Soliris (eculizumab) has not been studied for the treatment of acute relapses in NMOSD patients.
Paediatric population
Paroxysmal Nocturnal Haemoglobinuria
A total of 7 PNH paediatric patients, with a median weight of 57.2 kg (range of 48.6 to 69.8 kg) and aged from 11 to 17 years (median age : 15.6 years), received Soliris in study M07-005.
Treatment with eculizumab at the proposed dosing regimen in the paediatric population was associated with a reduction of intravascular haemolysis as measured by serum LDH level. It also resulted in a marked decrease or elimination of blood transfusions, and a trend towards an overall improvement in general function. The efficacy of eculizumab treatment in paediatric PNH patients appears to be consistent with that observed in adult PNH patients enrolled in PNH pivotal Studies (C04-001 and C04-002) (Table 3 and 14).
Table 14. Efficacy Outcomes in Paediatric PNH Study M07-005:
| P–Value | |||
|---|---|---|---|
| Mean (SD) | Wilcoxon Signed Rank | Paired t-test | |
| Change from baseline at 12 weeks of LDH Value (U/L) | -771 (914) | 0,0156 | 0,0336 |
| LDH AUC (U/L x Day)) | -60,634 (72,916) | 0,0156 | 0.0350 |
| Change from baseline at 12 weeks in Plasma Free Haemoglobin (mg/dL) | -10.3 (21.13) | 0.2188 | 0.1232 |
| Change from baseline Type III RBC clone size (Percent of aberrant cells) | 1.80 (358.1) | ||
| Change from baseline at 12 weeks of PedsQLTM 4.0 Generic Core scale (patients) | 10.5 (6.66) | 0.1250 | 0.0256 |
| Change from baseline at 12 weeks of PedsQLTM 4.0 Generic Core scale (parents) | 11.3 (8.5) | 0.2500 | 0.0737 |
| Change from baseline at 12 weeks of PedsQLTM Multidimensional Fatigue (patients) | 0.8 (21.39) | 0.6250 | 0.4687 |
| Change from baseline at 12 weeks of PedsQLTM Multidimensional Fatigue (parents) | 5.5 (0.71) | 0.5000 | 0.0289 |
Atypical Haemolytic Uremic Syndrome
A total of 15 paediatric patients (aged 2 months to 12 years) received Soliris in aHUS Study C09-001r. Forty seven percent of patients had an identified complement regulatory factor mutation or auto-antibody. The median time from aHUS diagnosis to first dose of Soliris was 14 months (range <1, 110 months). The median time from current thrombotic microangiopathy manifestation to first dose of Soliris was 1 month (range <1 to 16 months). The median duration of Soliris therapy was 16 weeks (range 4 to 70 weeks) for children <2 years of age (n=5) and 31 weeks (range 19 to 63 weeks) for children 2 to <12 years of age (n=10).
Overall, the efficacy results for these paediatric patients appeared consistent with what was observed in patients enrolled in aHUS pivotal Studies C08-002 and C08-003 (Table 6). No paediatric patient required new dialysis during treatment with Soliris.
Table 15. Efficacy Results in Paediatric Patients Enrolled in aHUS C09-001r:
| Efficacy Parameter | <2 years (n=5) | 2 to <12 years (n=10) | <12 years (n=15) |
|---|---|---|---|
| Patients with platelet count normalization, n (%) | 4 (80) | 10 (100) | 14 (93) |
| Complete TMA response, n (%) | 2 (40) | 5 (50) | 7 (50) |
| Daily TMA intervention rate, median (range) | |||
| Before eculizumab | 1 (0, 2) | <1 (0,07, 1,46) | <1 (0, 2) |
| On eculizumab treatment | <1 (0, <1) | 0 (0, <1) | 0 (0, <1) |
| Patients with eGFR improvement ≥15 mL/min/1,73 m², n (%) | 2 (40) | 6 (60) | 8 (53) |
In paediatric patients with shorter duration of current severe clinical thrombotic microangiopathy (TMA) manifestation prior to eculizumab, there was TMA control and improvement of renal function with eculizumab treatment (Table 15).
In paediatric patients with longer duration of current severe clinical TMA manifestation prior to eculizumab, there was TMA control with eculizumab treatment. However, renal function was not changed due to prior irreversible kidney damage (Table 16).
Table 16. Efficacy Outcomes in Paediatric Patients in Study C09-001r according to duration of current severe clinical thrombotic microangiopathy (TMA) manifestation:
| Duration of current severe clinical TMA manifestation | ||
|---|---|---|
| <2 months N=10 (%) | >2 months N=5 (%) | |
| Platelet count normalization | 9 (90) | 5 (100) |
| TMA event-free status | 8 (80) | 3 (60) |
| Complete TMA response | 7 (70) | 0 |
| eGFR improvement ≥15 mL/min/1,73 m² | 7 (70) | 0* |
* One patient achieved eGFR improvement after renal transplant
A total of 22 paediatric and adolescents patients (aged 5 months to 17 years) received Soliris in aHUS Study C10-003.
In Study C10-003, patients who enrolled in the study were required to have a platelet count < lower limit of normal range (LLN), evidence of haemolysis such as an elevation in serum LDH above the upper limits of normal and serum creatinine level ≥97 percentile for age without the need for chronic dialysis. The median patient age was 6.5 years (range: 5 months to 17 years). Patients enrolled in aHUS C10-003 had an ADAMTS-13 level above 5%. Fifty percent of patients had an identified complement regulatory factor mutation or auto-antibody. A total of 10 patients received PE/PI prior to eculizumab. Table 17 summarizes the key baseline clinical and disease-related characteristics of patients enrolled in aHUS Study C10-003.
Table 17. Baseline Characteristics of Paediatric and Adolescents Patients Enrolled in aHUS Study C10-003:
| Parameter | 1 month to <12 years (N=18) | All Patients (N=22) |
|---|---|---|
| Time from aHUS diagnosis until first study dose (months) median (min, max) | 0,51 (0,03, 58) | 0,56 (0,03, 191) |
| Time from current clinical TMA manifestation until first study dose (months), median (min, max) | 0,23 (0,03, 4) | 0,20 (0,03, 4) |
| Baseline platelet count (×109/L), median (min, max) | 110 (19, 146) | 91 (19, 146) |
| Baseline LDH (U/L) median (min, max) | 1510 (282, 7164) | 1244 (282, 7164) |
| Baseline eGFR (mL/min/1,73 m²), median (min, max) | 22 (10, 105) | 22 (10, 105) |
Patients in aHUS C10-003 received Soliris for a minimum of 26 weeks. After completion of the initial 26-week treatment period, most patients elected to continue on chronic dosing. Reduction in terminal complement activity was observed in all patients after commencement of Soliris. Soliris reduced signs of complement-mediated TMA activity, as shown by an increase in mean platelet counts from baseline to 26 weeks. The mean (±SD) platelet count increased from 88 ± 42 x1109/L at baseline to 281 ± 123 x109/L by one week; this effect was maintained through 26 weeks (mean platelet count (±SD) at week 26: 293 ± 106 x109/L). Renal function, as measured by eGFR, was improved during Soliris therapy. Nine of the 11 patients who required dialysis at baseline no longer required dialysis after Study Day 15 of eculizumab treatment. Responses were similar across all ages from 5 months to 17 years of age. In aHUS C10-003, responses to Soliris were similar in patients with and without identified mutations in genes encoding complement regulatory factor proteins or auto-antibodies to factor H.
Table 18 summarizes the efficacy results for aHUS C10-003.
Table 18. Efficacy Outcomes in Prospective aHUS Study C10-003:
| Efficacy Parameter | 1 month to <12 years (N=18) At 26-weeks | All Patients (N=22) At 26-weeks |
|---|---|---|
| Complete Hematologic Normalization, n (%) | 14 (78) | 18 (82) |
| Median Duration of complete hematologic normalization, weeks (range)1 | 35 (13, 78) | 35 (13, 78) |
| Complete TMA response, n (%) | 11 (61) | 14 (64) |
| Median Duration of complete TMA response, weeks (range)1 | 40 (13, 78) | 37 (13, 78) |
| TMA Event-Free Status, n (%) | 17 (94) | 21 (96) |
| 95% CI | NA | 77; 99 |
| Daily TMA Intervention rate, median (range) | ||
| Before eculizumab treatment, median | NA | 0,4 (0, 1.7) |
| On eculizumab treatment, median | NA | 0 (0, 1.01) |
| eGFR improvement ≥15 mL/min/1.73•m², n (%) | 16 (89) | 19 (86) |
| Change in eGFR (≥15 mL/min/1.73•m²) at 26 weeks, median (range) | 64 (0,146) | 58 (0, 146) |
| CKD improvement by ≥1 stage, n (%) | 14/16 (88) | 17/20 (85) |
| PE/PI Event-Free Status, n (%) | 16 (89) | 20 (91) |
| New Dialysis Event-Free Status, n (%) | 18 (100) | 22 (100) |
| 95% CI | NA | 85; 100 |
1 Through data cut-off (October 12, 2012), with median duration of Soliris therapy of 44 weeks (range: 1 dose to 88 weeks).
Longer term treatment with Soliris (median 55 weeks ranging from 1day to 107 weeks) was associated with an increased rate of clinically meaningful improvements in paediatric and adolescent patients with aHUS. When Soliris treatment was continued for more than 26 weeks, one additional patient (68% of patients in total) achieved Complete TMA Response and two additional patients (91% of patients in total) achieved hematologic normalization. At the last evaluation, 19 of 22 patients (86%) achieved eGFR improvement of ≥15 mL/min/1.73 m² from baseline. No patient required new dialysis with Soliris.
Refractory Generalized Myasthenia Gravis
A total of 11 paediatric patients with refractory gMG received Soliris in study ECU-MG-303. The median (range) body weight of the treated patients was 59.7 kg (37.2 to 91.2 kg) at baseline, and the median (range) age of 15 years (12 to 17 years) at screening. All patients included in the study were patients with refractory gMG who had one or more of the following:
1. Failed treatment ≥1 year with at least 1 IST, defined as: (i) Persistent weakness with impairment of activities of daily living, or (ii) Myasthenia gravis exacerbation and/or crisis while on treatment, or (iii) Intolerance to ISTs due to side effect or comorbid condition(s).
2. Require maintenance PE or IVIg to control symptoms (ie, patients who require PE or IVIg on a regular basis for the management of muscle weakness at least every 3 months over the last 12 months prior to screening).
The baseline characteristics of the paediatric patients with refractory gMG enrolled in study ECU-MG-303 are outlined in Table 19.
Table 19. Patient Demographic and Characteristics in Study ECU-MG-303:
| Eculizumab (n=11) | ||
|---|---|---|
| Female | n (%) | 9 (81.8%) |
"Duration of MG (time from MG
diagnosis to first study drug date
[years]) |Mean (SD)
Median (min, max) |3.99 (2.909)
2.90 (0.1, 8.8) |
|Baseline MG-ADL total score |Mean (SD)
Median (min, max) |5.0 (5.25)
4.0 (0, 19) |
|Baseline QMG total score |Mean (SD)
Median (min, max) |16.7 (5.64)
15.0 (10, 28) |
|MGFA classification at Screening
IIa
IIb
IIIa
IIIb
IVa
IVb | n (%)
|
2 (18.2)
3 (27.3)
3 (27.3)
0
3 (27.3)
0 |
|Patients with prior MG exacerbation
including MG crisis since diagnosis
No
Yes
Exacerbation
MG crisis | n (%)
|
4 (36.4)
7 (63.6)
6 (54.5)
3 (27.3)|
|Chronic IVIg therapy at study entry
Yes
No |n (%)
|
6 (54.5)
5 (45.5) |
|Number of immunosuppressant
therapies at Baseline
0
1
2 |n (%)
|
2 (18.2)
4 (36.4)
5 (45.5)|
|Patients with any immunosuppressant
therapiesa at Baseline n (%)
Corticosteroids
Azathioprine
Mycophenolate mofetil
Tacrolimus|n (%)
|
8 (72.7)
1 (9.1)
2 (18.2)
3 (27.3) |
a Immunosuppressant therapies included corticosteroids, azathioprine, cyclophosphamide, cyclosporine, methotrexate, mycophenolate mofetil, or tacrolimus. No patient received cyclosporine, cyclophosphamide, or methotrexate at Baseline.
Abbreviations: IVIg = intravenous immunoglobulin; max = maximum; MG = myasthenia gravis; MG-ADL = Myasthenia Gravis Activities of Daily Living profile; MGFA = Myasthenia Gravis Foundation of America; min = minimum; QMG = Quantitative Myasthenia Gravis score for disease severity; SD = standard deviation
The primary endpoint of study ECU-MG-303 was the change from baseline in the QMG total score over time regardless of rescue therapy. Paediatric patients treated with Soliris demonstrated a statistically significant improvement from baseline in QMG total score throughout the Primary Evaluation Treatment Period of 26 weeks. The results for the primary and main secondary endpoint in study ECU-MG-303 are included in Table 20.
The efficacy of Soliris treatment in paediatric patients with refractory gMG was consistent with that observed in adult patients with refractory gMG enrolled in the pivotal study ECU-MG-301 (Table 10).
Table 20. Efficacy Outcomes in Study ECU-MG-303:
| Efficacy Endpoints: Total Score Change from Baseline at Week 26 | LS Mean (SEM) 95% CI |
|---|---|
| QMG | -5.8 (1.2) (-8.40, -3.13) na = 10 |
| MG-ADL total score | -2.3 (0.6) (-3.63, -1.03) na = 10 |
| MGC total score | -8.8 (1.9) (-12.93, -4.69) na = 9 |
a n is the number of patients at Week 26
Abbreviations: CI = confidence interval; LS = least squares; MG-ADL = Myasthenia Gravis Activities of Daily Living profile; MGC = Myasthenia Gravis Composite; QMG = Quantitative Myasthenia Gravis score for disease severity; SEM = standard error of mean; VAS = visual analog scale
In study ECU-MG-303, a clinical responder in the QMG and MG-ADL total scores was defined as having at least a 5-point improvement and 3-point improvement from baseline, respectively. The proportion of clinical responders in the QMG and MG-ADL total scores at Week 26 regardless of rescue therapy was 70% and 50%, respectively. The 10 patients who completed their visit at Week 26 achieved improved status of MGFA Post-Interventional Status (MGFA-PIS) at Week 26. Seven (70%) patients achieved minimal manifestation of refractory gMG at Week 26.
An event of clinical deterioration (MG crisis) was observed in 1 patient (9.1%) during the Primary Evaluation Treatment Period requiring rescue therapy (PE) which was administered between the Week 22 and Week 24 study visits. As a result and due to physician decision, this patient did not have QMG, MG-ADL or other efficacy assessments after Week 20 and did not enter the extension period.
During the Primary Evaluation Treatment Period in paediatric patients with refractory gMG (study ECU-MG-303), 1 out of 11 patients (9.1%) decreased daily dose of anticholinesterase and 3 out of 11 patients (27.3%) decreased their daily dose of corticosteroid, due to improved MG symptoms.
Neuromyelitis Optica Spectrum Disorder
Soliris has not been evaluated in paediatric patients with NMOSD.
The European Medicines Agency has deferred the obligation to submit the results of studies with Soliris in one or more subsets of the paediatric population in the treatment of NMOSD (see section 4.2 for information on paediatric use).
Pharmacokinetic properties
Pharmacokinetics and Drug Metabolism
Biotransformation
Human antibodies undergo endocytotic digestion in the cells of the reticuloendothelial system. Eculizumab contains only naturally occurring amino acids and has no known active metabolites. Human antibodies are predominately catabolized by lysosomal enzymes to small peptides and amino acids.
Elimination
No specific studies have been performed to evaluate the hepatic, renal, lung, or gastrointestinal routes of excretion/elimination for Soliris. In normal kidneys, antibodies are not excreted and are excluded from filtration by their size.
Pharmacokinetic Parameters
In 40 patients with PNH, a 1-compartmental model was used to estimate pharmacokinetic parameters after multiple doses. Mean clearance was 0.31 ± 0.12 mL/hr/kg, mean volume of distribution was 110.3 ± 17.9 mL/kg, and mean elimination half-life was 11.3 ± 3.4 days. The steady state is achieved by 4 weeks using the PNH adult dosing regimen.
In PNH patients, pharmacodynamic activity correlates directly with eculizumab serum concentrations and maintenance of trough levels above ≥35 microgram/mL results in essentially complete blockade of haemolytic activity in the majority of PNH patients.
A second population PK analysis with a standard 1 compartmental model was conducted on the multiple dose PK data from 37 aHUS patients receiving the recommended Soliris regimen in studies C08-002A/B and C08-003A/B. In this model, the clearance of Soliris for a typical aHUS patient weighing 70 kg was 0.0139 L/hr and the volume of distribution was 5.6 L. The elimination half-life was 297 h (approximately 12.4 days).
The second population PK model was applied to the multiple dose PK data from 22 paediatric aHUS patients receiving the recommended Soliris regimen in aHUS C10-003. The clearance and volume of distribution of Soliris are weight dependent, which forms the basis for a weight categorical based dose regimen in paediatric patients (see section 4.2). Clearance values of Soliris in paediatric aHUS patients were 10.4, 5.3, and 2.2 mL/hr with body weight of 70, 30, and 10 kg, respectively; and the corresponding volume of distribution values were 5.23, 2.76, and 1.21 L, respectively. The corresponding elimination half-life remained almost unchanged within a range of 349 to 378 h (approximately 14.5 to 15.8 days).
The clearance and half-life of eculizumab were also evaluated during plasma exchange interventions. Plasma exchange resulted in an approximately 50% decline in eculizumab concentrations following a 1 hour intervention and the elimination half-life of eculizumab was reduced to 1.3 hours. Supplemental dosing is recommended when Soliris is administered to aHUS patients receiving plasma infusion or exchange (see section 4.2).
All aHUS patients treated with Soliris when administered as recommended demonstrated rapid and sustained reduction in terminal complement activity. In aHUS patients, pharmacodynamic activity correlates directly with eculizumab serum concentrations and maintenance of trough levels of approximately 50-100 microgram/ml results in essentially complete blockade of terminal complement activity in all aHUS patients.
PK parameters are consistent across PNH, aHUS, refractory gMG and NMOSD patient populations. Pharmacodynamic activity measured by free C5 concentrations of <0.5 ug/mL, is correlated with essentially complete blockade of terminal complement activity in PNH, aHUS, refractory gMG and NMOSD patients.
Special Populations
Dedicated studies have not been conducted to evaluate the pharmacokinetics of Soliris in special patient populations identified by gender, race, age (geriatric), or the presence of renal or hepatic impairment. Population PK analysis on data collected across studies in PNH, aHUS, gMG and NMOSD patients showed that gender, race, age (geriatric), or the presence of renal or hepatic impairment function do not influence the PK of eculizumab. Body weight was a significant covariate resulting in a lower eculizumab clearance in paediatric patients requiring body weight based dosing in paediatric patients.
Paediatric population
The pharmacokinetics of eculizumab was evaluated in Study M07-005 in PNH paediatric patients (aged from 11 to less than 18 years), in Studies C08-002, C08-003, C09-001r and C10-003 in aHUS paediatric patients (aged 2 months to less than 18 years), and in Study ECU-MG-303 paediatric patients with refractory gMG (aged from 12 years to less than 18 years) with body-weight based dose regimen. Weight was a significant covariate resulting in a lower eculizumab clearance 0.0105 L/h in the adolescent PNH patients.
Preclinical safety data
The specificity of eculizumab for C5 in human serum was evaluated in two in vitro studies.
The tissue cross-reactivity of eculizumab was evaluated by assessing binding to a panel of 38 human tissues. C5 expression in the human tissue panel examined in this study is consistent with published reports of C5 expression, as C5 has been reported in smooth muscle, striated muscle, and renal proximal tubular epithelium. No unexpected tissue cross-reactivity was observed.
Animal reproduction studies have not been conducted with eculizumab due to lack of pharmacologic activity in non-human species.
In a 26 week toxicity study performed in mice with a surrogate antibody directed against murine C5, treatment did not affect any of the toxicity parameters examined. Haemolytic activity during the course of the study was effectively blocked in both female and male mice.
No clear treatment-related effects or adverse effects were observed in reproductive toxicology studies in mice with a surrogate terminal complement inhibitory antibody, which was utilized to assess the reproductive safety of C5 blockade. These studies included assessment of fertility and early embryonic development, developmental toxicity, and pre and post-natal development.
When maternal exposure to the antibody occurred during organogenesis, two cases of retinal dysplasia and one case of umbilical hernia were observed among 230 offspring born to mothers exposed to the higher antibody dose (approximately 4 times the maximum recommended human Soliris dose, based on a body weight comparison); however, the exposure did not increase foetal loss or neonatal death.
No animal studies have been conducted to evaluate the genotoxic and carcinogenic potential of eculizumab.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.