SOTYKTU Film-coated tablet Ref.[50886] Active ingredients: Deucravacitinib
Source: European Medicines Agency (EU) Revision Year: 2023 Publisher: Bristol-Myers Squibb Pharma EEIG, Plaza 254, Blanchardstown Corporate Park 2, Dublin 15, D15 T867, Ireland
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Immunosuppressants, selective immunosuppressant
ATC code: L04AA56
Mechanism of action
Deucravacitinib selectively inhibits the TYK2 enzyme (TYK2 belongs to the JAK family). Deucravacitinib binds to the regulatory domain of TYK2, stabilizing an inhibitory interaction between the regulatory and the catalytic domains of the enzyme. This results in allosteric inhibition of receptormediated activation of TYK2 and its downstream functions in cells. TYK2 mediates signalling of interleukin-23 (IL-23), interleukin-12 (IL-12), and type I interferons (IFN), which are naturally occurring cytokines involved in inflammatory and immune responses. Deucravacitinib inhibits the release of proinflammatory cytokines and chemokines.
Pharmacodynamic effects
In patients with psoriasis, deucravacitinib reduced psoriasis associated gene expression in psoriatic skin including reductions in IL-23-pathway and type I IFN pathway regulated genes. Deucravacitinib reduced IL-17A, IL-19 and β-defensin by 47-50%, 72% and 81-84%, respectively following 16 weeks of once daily treatment.
Clinical efficacy and safety
The efficacy and safety of deucravacitinib were assessed in two multicentre, randomised, double-blind, placebo- and apremilast-controlled clinical studies (POETYK PSO-1 and POETYK PSO-2) in patients who were 18 years of age and older with moderate-to-severe plaque psoriasis and were eligible for systemic therapy or phototherapy. Patients had body surface area (BSA) involvement of ≥ 10%, a Psoriasis Area and Severity Index (PASI) score ≥ 12, and a static Physician's Global Assessment (sPGA) ≥ 3 (moderate or severe) on a 5-point scale of overall disease severity.
POETYK PSO-1 and POETYK PSO-2 evaluated a total of 1686 patients with 843 randomised to deucravacitinib 6 mg once daily, 422 to apremilast 30 mg twice daily, and 421 to placebo.
In both studies, patients receiving placebo switched to deucravacitinib at week 16, which continued up to week 52. Patients randomised to apremilast who did not achieve a PASI 50 (POETYK PSO-1) or PASI 75 (POETYK PSO-2) response at week 24 switched to deucravacitinib, and continued up to week 52. In POETYK PSO-1 patients who were randomised to deucravacitinib continued treatment up to week 52. In POETYK PSO-2, deucravacitinib treated patients who achieved PASI 75 at week 24 were re-randomised 1:1 to continue deucravacitinib (maintenance) or were switched to placebo (withdrawal).
Baseline disease characteristics were consistent for the study population in both studies: the majority of patients were male (67%), mean age was approximately 47 years old with the majority of patients between 40 and 64 years of age. 10% of patients were ≥ 65 years of age. The overall median PASI score was 18.7, and median BSA was 20%. Baseline sPGA score was 3 (moderate) in 79.8% of patients and 4 (severe) in 20.2%. Median Dermatology Life Quality Index (DLQI) score was 11. A total of 18.4% of study patients had a history of psoriatic arthritis.
Across both studies, 40% of patients had received prior phototherapy, 42.4% were naive to any systemic therapy (including biologic and/or non-biologic treatment), 41% received prior non-biologic systemic treatment, and 34.8% had received prior biologic therapy (16.1% TNF, 4.9% IL-12/23, 16.6% IL-17 and 4.4% IL-23 inhibitors).
The co-primary endpoints in the two studies were the proportions of patients who achieved 1) at least a 75% improvement in PASI scores (PASI 75) from baseline and 2) a sPGA score of clear or almost clear (0 or 1) at week 16 versus placebo.
In study POETYK PSO-1, PASI 75 was achieved with deucravacitinib in 58.4%, with apremilast in 35.1% and with placebo in 12.7% of the patients at week 16. Static Physician's Global Assessment (sPGA) of clear or almost clear at week 16 was achieved in 53.6%, 32.1% and 7.2% of the patients in the deucravacitinib, apremilast and placebo groups respectively. For these co-primary endpoints superiority of deucravacitinib to placebo was demonstrated. Consistent results were seen in study POETYK PSO-2.
Table 2 presents the main efficacy results for the co-primary and other endpoints.
Table 2. Main efficacy results in adults with plaque psoriasis:
| POETYK PSO-1 | POETYK PSO-2 | |||||
|---|---|---|---|---|---|---|
| Endpoint | Deucravacitinib (N=332) n (%) | Apremilast (N=168) n (%) | Placebo (N=166) n (%) | Deucravacitinib (N=511) n (%) | Apremilast (N=254) n (%) | Placebo (N=255) n (%) |
| sPGA 0/1 | ||||||
| Week 16 | 178 (53.6) | 54 (32.1)d | 12 (7.2)a,d | 253 (49.5) | 86 (33.9)d | 22 (8.6)a,d |
| Week 24 | 195 (58.7) | 52 (31.0)d | - | 251 (49.8)b | 75 (29.5)d | - |
| sPGA 0 | ||||||
| Week 16 | 58 (17.5) | 8 (4.8)d | 1 (0.6)d | 80 (15.7) | 16 (6.3)e | 3 (1.2)d |
| PASI 75 | ||||||
| Week 16 | 194 (58.4) | 59 (35.1)d | 21 (12.7)a,d | 271 (53.0) | 101 (39.8)e | 24 (9.4)a,d |
| Week 24 | 230 (69.3) | 64 (38.1)d | - | 296 (58.7)b | 96 (37.8)d | - |
| PASI 90 | ||||||
| Week 16 | 118 (35.5) | 33 (19.6)e | 7 (4.2)d | 138 (27.0) | 46 (18.1)f | 7 (2.7)d |
| Week 24 | 140 (42.2) | 37 (22.0)d | - | 164 (32.5)b | 50 (19.7)d | - |
| PASI 100 | ||||||
| Week 16 | 47 (14.2) | 5 (3.0)d | 1 (0.6)d | 52 (10.2) | 11 (4.3)f | 3 (1.2)d |
| Scalp Specific PGA 0/1c | (N=209) | (N=110) | (N=121) | (N=305) | (N=166) | (N=173) |
| Week 16 | 147 (70.3) | 43 (39.1)d | 21 (17.4)d | 182 (59.7) | 61 (36.7)d | 30 (17.3)d |
Non-responder imputation (NRI) was used; patients who discontinued treatment or the study prior to the endpoint or had missing data were counted as non-responders.
a Co-primary endpoint comparing deucravacitinib with placebo
b N=504 accounting for missed assessments due to COVID-19 pandemic
c Includes patients with baseline Scalp Specific PGA score of ≥ 3
d p≤0.0001 for comparison between deucravacitinib and placebo or deucravacitinib and apremilast
e p<0.001 for comparison between deucravacitinib and apremilast
f p<0.01 for comparison between deucravacitinib and apremilast
Examination of age, gender, race, body weight, duration of disease, baseline disease severity, and previous treatment with biologic or non-biologic agents did not identify differences in response to deucravacitinib among these subgroups.
Response over time
Deucravacitinib showed rapid onset of efficacy with maximum PASI 75 response achieved by week 24 (POETYK PSO-1 and PSO-2) and maintained through week 52 (POETYK PSO-1) (see Figure 1).
Figure 1. PASI 75 response (NRI) through week 52 by visit in POETYK PSO-1:
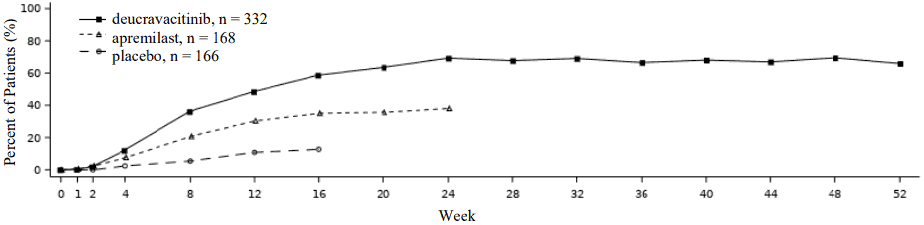
Maintenance and durability of response
In POETYK PSO-2, to evaluate maintenance and durability of response, patients who were originally randomised to deucravacitinib and achieved PASI 75 response at week 24, were re-randomised to either continue treatment on deucravacitinib or receive placebo. For responders at week 24 who were re-randomised to placebo, the median time to loss of PASI 75 response was approximately 12 weeks. Figure 2 shows the PASI 75 responses in the two arms from week 24-52.
Figure 2. PASI 75 response (NRI) after re-randomisation at week-24 in POETYK PSO-2:
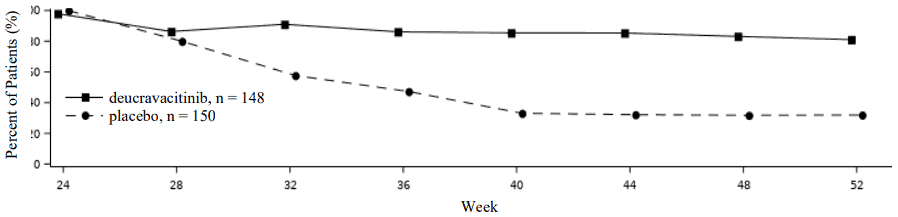
Patient reported outcomes
Significantly greater improvements in health-related quality of life as measured by Dermatology Life Quality Index (DLQI) and in patient reported psoriasis symptoms (itch, pain, burning, stinging, and skin tightness) and signs (skin dryness, cracking, scaling, shedding or flaking, redness, and bleeding) as measured by the Psoriasis Symptoms and Signs Diary (PSSD) were observed in deucravacitinibtreated patients compared to placebo at week 16 and to apremilast at week 16 and week 24. Improvement of these responses in patients receiving continuous deucravacitinib treatment were maintained through week 52 in POETYK PSO-1.
Table 3. Patient reported outcomes in POETYK PSO-1 and POETYK PSO-2:
| POETYK PSO-1 | POETYK PSO-2 | |||||
|---|---|---|---|---|---|---|
| Deucravacitinib | Apremilast | Placebo | Deucravacitinib | Apremilast | Placebo | |
| DLQI Patients achieving 0 or 1 (NRI)* | N=322 | N=161 | N=160 | N=495 | N=247 | N=246 |
| Week 16, n (%) | 132 (41.0) | 46 (28.6)a | 17 (10.6)b | 186 (37.6) | 57 (23.1)b | 24 (9.8)b |
| Week 24, n (%) | 155 (48.1) | 39 (24.2)b | - | 205 (41.4) | 53 (21.5)b | - |
| PSSD symptom score Change from baseline (mBOCF)** | N=306 | N=158 | N=151 | N=466 | N=233 | N=239 |
| Week 16, mean (SE) | -26.7 (1.8) | -17.8 (2.2)b | -3.6 (2.1)b | -28.3 (1.1) | -21.1 (1.4)b | -4.7 (1.4)b |
| Week 24, mean (SE) | -31.9 (2.0) | -20.7 (2.4)b | - | -29.1 (1.1) | -21.4 (1.5)b | - |
| PSSD sign score Change from baseline (mBOCF)* | N=306 | N=158 | N=151 | N=466 | N=233 | N=239 |
| Week 16, mean (SE) | -28.9 (1.8) | -20.0 (2.2)b | -5.3 (2.1)a | -31.9 (1) | -23.8 (1.4)b | -7.1 (1.4)b |
| Week 24, mean (SE) | -33.8 (2.0) | -22.5 (2.4)b | - | -32.4 (1.1) | -24.2 (1.5)b | - |
* Patients with baseline score ≥ 2
** Adjusted mean change; mBOCF – modified baseline observation carried forward; standard error (SE)
a p<0.01 for comparison between deucravacitinib and placebo or deucravacitinib and apremilast
b p<0.0001 for comparison between deucravacitinib and placebo or deucravacitinib and apremilast
Elderly population
Of the 1519 patients with plaque psoriasis treated with deucravacitinib in clinical studies, 152 patients were 65 years or older, including 21 patients who were 75 years or older (see section 4.2). No overall differences in exposure, safety or effectiveness were observed between older and younger patients who received deucravacitinib.
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with SOTYKTU in one or more subsets of the paediatric population in the treatment of psoriasis (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Deucravacitinib exhibited near complete oral absorption, dose-related increase in exposure, and no evident time-dependent pharmacokinetics.
Absorption
Following oral administration of tablets, deucravacitinib exhibited rapid and near complete absorption. The median Tmax ranged from 2 to 3 hours and absolute oral bioavailability was 99% in healthy volunteers. Modest accumulation (<1.4-fold at steady state) was observed following once daily dosing.
Food
Deucravacitinib can be administered without consideration for food or gastric pH modulators (H2 receptor blockers and proton pump inhibitors). Co-administration of food or gastric pH modulators did not affect total exposure (AUC[INF]) of deucravacitinib.
Distribution
The volume of distribution at steady state (Vss), is 140 L, which is greater than total body water [42 L] indicating extravascular distribution. Deucravacitinib is 81.6% bound to human plasma proteins, primarily to human serum albumin.
Deucravacitinib distributes similarly between plasma and red blood cell components with blood-toplasma concentration ratio of 1.26.
Biotransformation
In humans, deucravacitinib is metabolised via four primary biotransformation pathways, which include N-demethylation at the triazole moiety by cytochrome P-450 (CYP) 1A2 to form major metabolite BMT-153261, cyclopropyl carboxamide hydrolysis by carboxylesterase 2 (CES2) to form major metabolite BMT-158170, N-glucuronidation by uridine glucuronyl transferase (UGT) to form BMT-334616, and mono-oxidation by CYP 2B6/2D6 at the deuterated methyl group to form M11.
At steady state, deucravacitinib is the major circulating species constituting 49% of measured compound related components. Two major circulating metabolites, BMT-153261 and BMT-158170, were identified, both of which have half-lives comparable to the parent deucravacitinib. BMT-153261 has comparable potency to the parent compound and BMT-158170 is not pharmacologically active. The circulating exposure of BMT-153261 is much lower than the parent compound and therefore, the predominant pharmacological activity is attributed to the parent compound deucravacitinib.
Additionally, no unique to human metabolites and no long-lived circulatory metabolites were identified.
Elimination
Deucravacitinib is eliminated via multiple pathways, including Phase I and II metabolism, along with direct renal and faecal elimination. Additionally, no single enzyme contributed more than 26% of total clearance. Deucravacitinib is extensively metabolised, with 59% of orally administered [14C]-deucravacitinib dose eliminated as metabolites in urine (37% of the dose) and faeces (22% of the dose). Unchanged deucravacitinib in urine and faeces represented 13% and 26% of the dose, respectively.
The terminal elimination half-life of deucravacitinib 6 mg in healthy human adults is 10 hours, with a total clearance of 15.3 L/h (CV 27%). Deucravacitinib is a substrate of efflux transporters, P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP) and uptake transporter OCT1. Due to high passive permeability, high oral bioavailability and low affinity for these transporters, contribution of these transporters to deucravacitinib pharmacokinetics is minimal.
Deucravacitinib is not a substrate of transporters OATP, NTCP, OAT1, OAT3, OCT2, MATE1, or MATE2K.
Linearity/non-linearity
The pharmacokinetics of single doses of deucravacitinib administered as tablets was linear across 3 mg to 36 mg dose range.
Interactions
Effect of deucravacitinib on other medicinal products
In vitro studies have shown no evidence that deucravacitinib and its major circulating metabolites, at clinically relevant exposures, inhibit major CYPs (1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 3A4), UGTs (1A1, 1A4, 1A6, 1A9, 2B7), CES2 and drug transporters (P-gp, BCRP, OATP1B1, OATP1B3, BSEP, MRP2, OAT1, OAT3, OCT1, OCT2, MATE1, and MATE2K). Additionally, deucravacitinib does not induce CYP 1A2, 2B6, and 3A4 (see section 4.5).
Special populations
Elderly
Based on the population pharmacokinetic analysis, deucravacitinib mean steady state exposure (Cavg,ss) was higher, 31% in patients aged 65-74 years [n=87 of 1387 (6.3%)] and 53% in patients aged 75-84 years [n=13 of 1387 (0.94%)]. Exposures in patients aged ≥85 years old are not available.
Patients with renal impairment
Renal impairment has no clinically meaningful effect on deucravacitinib exposures (see section 4.2) based on a dedicated study where estimated glomerular filtration rate (eGFR) was determined using a modification of diet in renal disease (MDRD) equation. Compared to normal renal function group, deucravacitinib Cmax was altered by up to 15% and AUC[INF] increased by up to 48% across renal impairment groups (mild (eGFR: ≥60 to <90 mL/min), moderate (eGFR: ≥30 to <60 mL/min), severe (eGFR: <30 mL/min), and ESRD (eGFR: <15 mL/min)). Compared to the normal renal function group, BMT-153261 Cmax increased by up to 34% and AUC[INF] increased up to 84% across renal impairment groups.
Dialysis does not substantially clear deucravacitinib from systemic circulation (5.4% of dose cleared per dialysis).
Patients with hepatic impairment
Mild (Child-Pugh Class A) and moderate (Child-Pugh Class B) hepatic impairment has no clinically meaningful effect on deucravacitinib exposures (see section 4.2). Compared to normal hepatic function group, total deucravacitinib Cmax and AUC[INF] in mild and moderate hepatic impairment group increased by up to 10% and 40%, respectively while the unbound deucravacitinib Cmax and AUC(INF) increased by up to 26% and 60%, respectively. In severe (Child-Pugh Class C) hepatic impaired adults, total deucravacitinib Cmax was comparable and total AUC was 43% higher relative to matched healthy adults. In these adults, unbound Cmax and AUC(INF) increased by 62% and 131%, respectively. Deucravacitinib is not recommended for use in patients with severe hepatic impairment (see section 4.2).
The AUC(0-T) of BMT-153261 decreased by 19%, 53% and 76% in subjects with mild, moderate, and severe hepatic impairment, respectively, compared to subjects with normal hepatic function, while Cmax of BMT-153261, decreased by 25%, 59%, and 79% in subjects with mild, moderate, and severe hepatic impairment, respectively.
Gender
Based on population pharmacokinetic modelling and simulation, females are expected to have an about 30% higher deucravacitinib mean steady-state exposure (Cmax,ss and Cavg,ss) compared to male.
Body weight
Based on population pharmacokinetic modelling and simulation, patients with lower body weight (<60 kg) are expected to have a higher geometric mean steady-state exposure of deucravacitinib of 37.4% (Cmaxss) and 24.8% (Cavgss). Patients with a higher body weight (>90 kg) are expected to have a lower geometric mean steady-state deucravacitinib exposure of 24.8% (Cmax,ss) and 19.6% (Cavgss) (compared to patients with body weight 60-90 kg).
Intrinsic factors
Race, and ethnicity did not have a clinically meaningful effect on deucravacitinib exposure.
5.3. Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential, and toxicity to reproduction and development.
Repeated dose toxicity
In the chronic toxicity study in rats, decreases in lymphocyte counts, bone marrow cellularity and lymphoid cellularity in tissues of the immune system were observed at exposure (AUC) at lowestobserved-effect-level (LOEL) approximately 9 times the recommended human dose (RHD). These effects were not associated with clinical signs of immunosuppression (e.g., infections). Decreases in platelet counts and red blood cell (RBC) mass parameters were observed at exposure (AUC) at the LOEL approximately 42 times the RHD. In the chronic toxicity study in monkeys, clinical and microscopic skin changes and decreased RBC mass parameters were observed at exposure (AUC) at LOEL approximately 7 times the RHD.
Developmental and reproductive toxicity
Deucravacitinib had no effects on fertility or early embryonic development in male and female rats at exposures (AUC) up to approximately 247 and 171 times the RHD, respectively. Deucravacitinib was neither embryo-lethal nor teratogenic at maternal exposures (AUC) up to approximately 266 times the RHD in rats or 91/20 (total/free) times the RHD in rabbits.
In a pre- and post-natal development study in rats, transiently lower pup body weights were noted during the pre-weaning period at maternal exposure (AUC) approximately 110 times the RHD. This effect fully recovered during the post-weaning period.
Following administration of radiolabelled deucravacitinib to lactating rats, deucravacitinib and/or its metabolites were present in the milk with milk-to-plasma concentration ratios of 2.7 to 30.9.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.