STAQUIS Ointment Ref.[28063] Active ingredients: Crisaborole
Source: European Medicines Agency (EU) Revision Year: 2021 Publisher: Pfizer Europe MA EEIG, Boulevard de la Plaine 17, 1050 Bruxelles, Belgium
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Other dermatological preparations, agents for dermatitis, excluding corticosteroids
ATC code: D11AH06
Mechanism of action
Crisaborole is an anti-inflammatory benzoxaborole phosphodiesterase-4 (PDE4) inhibitor that suppresses secretion of certain cytokines, such as tumour necrosis factor-α (TNF-α), interleukins (IL-2, IL-4, IL-5), and interferon gamma (IFNγ), and improves skin barrier function as measured by transepidermal water loss (TEWL). Crisaborole applied on atopic dermatitis lesions of patients reduces expression of atopic inflammation associated chemokines including CCL17, CCL18, and CCL22.
Clinical efficacy and safety
Two multicentre, randomised, double-blind, parallel-group, vehicle-controlled trials (Trials 1 and 2), identical in design, included a total of 1,522 subjects 2 to 79 years of age. 61.9% of subjects were 2-11 years old, 24.4% of subjects were 12-17 years old, 13.3% of subjects were 18-64 years old, and 0.5% of subjects were 65 years of age or older; the number of subjects ≥18 years of age was limited. The treatable BSA ranged from 5% to 95% (mean = 18.3%, standard deviation [SD] = 17.8%; 9.6% of subjects had >40% treatable BSA); the trials did not include sufficient numbers of subjects with >40% treatable BSA to determine the safety and efficacy of Staquis in this subpopulation. At baseline (pooled study data), 38.5% of the subjects had an Investigator’s Static Global Assessment (ISGA) score of 2 (Mild), and 61.5% had an ISGA score of 3 (Moderate), in the overall assessment of atopic dermatitis (erythema, induration/papulation, and oozing/crusting) on a severity scale of 0 to 4.
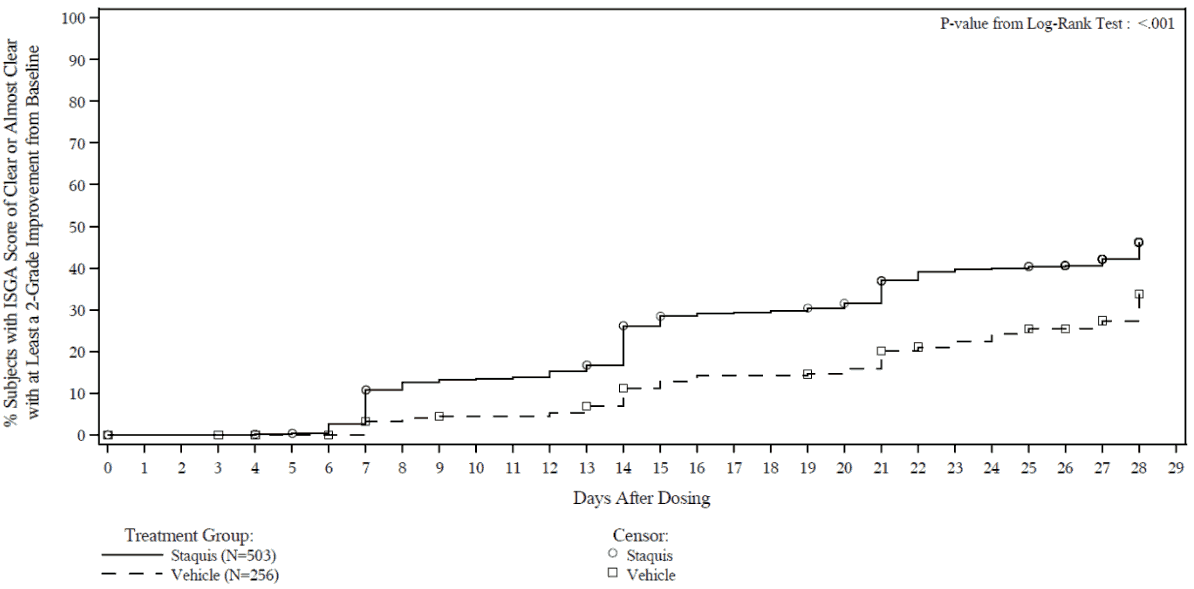
In both trials, subjects were randomised 2:1 to receive Staquis or vehicle applied twice daily for 28 days. The primary efficacy endpoint was the proportion of subjects at Day 29 who achieved an ISGA grade of Clear (score of 0) or Almost Clear (score of 1) with at least a 2-grade improvement from baseline, comparing Staquis-treated subjects to vehicle-treated subjects. In both trials, a statistically significantly greater percentage of subjects achieved this endpoint in the Staquis-treated group compared with the vehicle-treated group.
The secondary efficacy endpoints were the proportion of subjects at Day 29 with an ISGA grade of Clear or Almost Clear and the time to achieve an ISGA grade of Clear or Almost Clear with at least a 2-grade improvement from baseline.
The safety and efficacy of Staquis on sensitive skin areas (such as the face and neck) compared to nonsensitive skin areas (such as the arms and legs) were not separately assessed in the clinical trials.
Efficacy results from the two trials are summarised in Tables 2 and 3. The Kaplan-Meier plots for the time to achieve an ISGA score of Clear or Almost Clear with at least a 2-grade improvement from baseline are provided in Figures 1 and 2. The log-rank test p-values for both trials were <0.001.
Table 2. Efficacy outcomes in subjects with mild to moderate atopic dermatitis:
| Trial 1 | Trial 2 | |||
|---|---|---|---|---|
| Staquis (N=503) | Vehicle (N=256) | Staquis (N=513) | Vehicle (N=250) | |
| ISGA score of Clear or Almost Clear with at least a 2-grade improvement from baseline at Day 29 | 32.8% | 25.4% | 31.4% | 18.0% |
| 95% CIa | (28.6, 37.0) | (19.9, 30.9) | (27.3, 35.5) | (13.2, 22.9) |
| p-value | 0.038b | <0.001b | ||
| ISGA of Clear or Almost Clear at Day 29 | 51.7% | 40.6% | 48.5% | 29.7% |
| 95% CΙa | (47.2, 56.1) | (34.4, 46.8) | (44.1, 52.9) | (23.9, 35.5) |
| p-value | 0.005b | <0.001b | ||
a Confidence Interval (CI) from normal approximation.
b p-value from a logistic regression (with Firth option) test with factors of treatment group and analysis centre after adjusted for multiple imputation.
Table 3. Post-hoc efficacy outcomes in subjects with mild to moderate atopic dermatitis with ≤40% BSA affected:
| Trial 1 | Trial 2 | |||
|---|---|---|---|---|
| Staquis (N=446) | Vehicle (N=231) | Staquis (N=465) | Vehicle (N=234) | |
| ISGA score of Clear or Almost Clear with at least a 2-grade improvement from baseline at Day 29 | 34.1% | 25.5% | 32.6% | 18.8% |
| 95% CIa | (29.7, 38.6) | (19.7, 31.3) | (28.3, 36.9) | (13.7, 24.0) |
| p-value | 0.022b | <0.0001b | ||
| ISGA of Clear or Almost Clear at Day 29 | 53.8% | 41.9% | 51.0% | 30.9% |
| 95% CIa | (49.1, 58.5) | (35.3, 48.4) | (46.4, 55.6) | (24.8, 37.0) |
| p-value | 0.0041b | <0.0001b | ||
a Confidence Interval (CI) from normal approximation.
b p-value from a logistic regression (with Firth option) test with factors of treatment group and analysis centre after adjusted for multiple imputation.
Figure 1. Kaplan-Meier plot of Time to ISGA score of Clear or Almost Clear with at least a 2-grade improvement from baseline for subjects with mild to moderate atopic dermatitis:
Trial 1:
Trial 2:
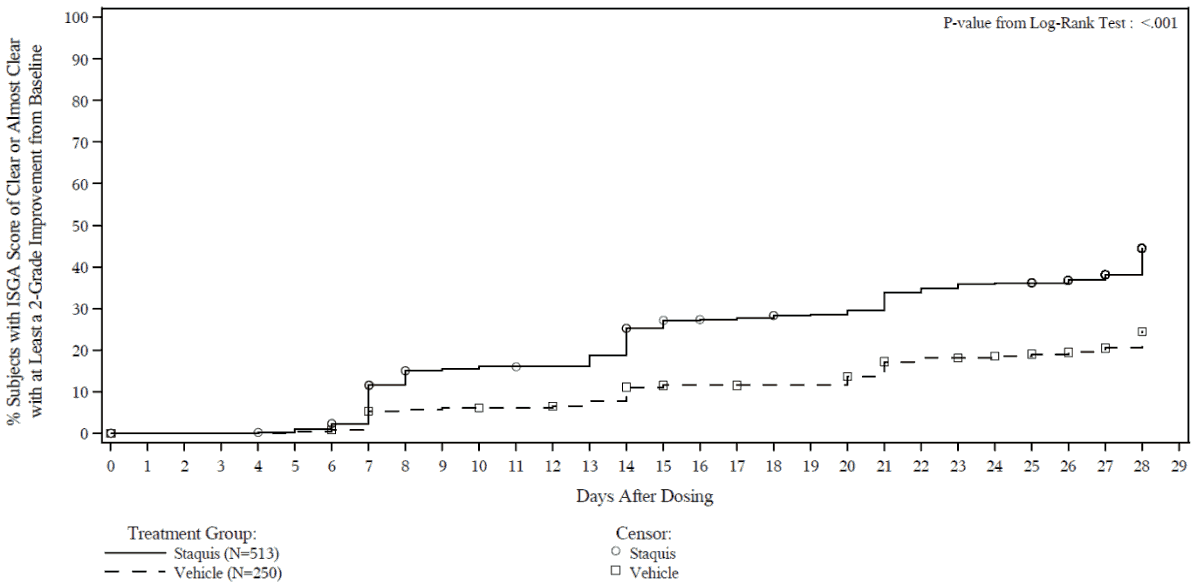
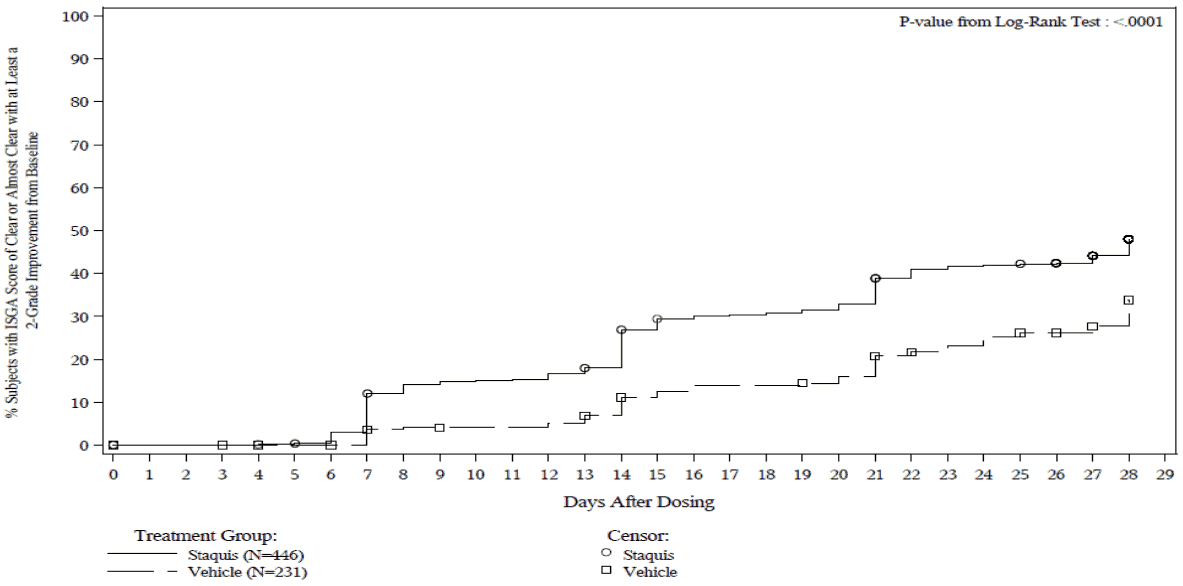
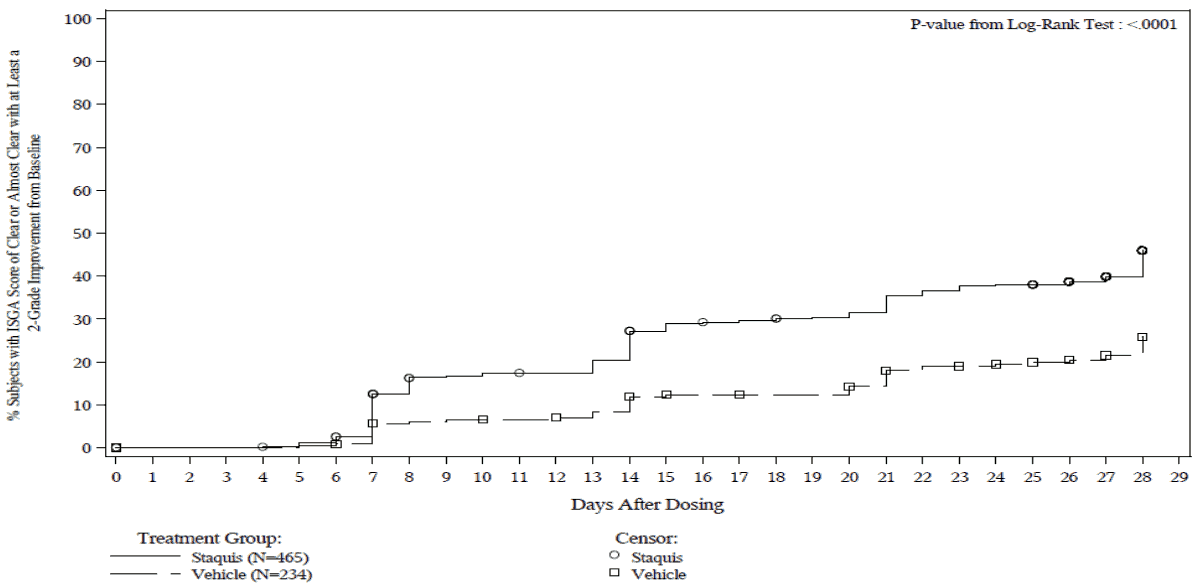
Figure 2. Post-hoc Kaplan-Meier plot of Time to ISGA score of Clear or Almost Clear with at least a 2-grade improvement from baseline for subjects with mild to moderate atopic dermatitis with ≤40% BSA affected:
Trial 1:
Trial 2:
The pooled primary efficacy results by race category are summarised in Table 4.
Table 4. Summary of subjects achieving ISGA score of Clear or Almost Clear with at least a 2-grade improvement from baseline at Day 29 by race category – Trial 1 and 2 pooled:
| Staquis (N=1016) | Vehicle (N=506) | |||
|---|---|---|---|---|
| Race Category | n | Rate | n | Rate |
| American Indian or Alaska Native | 11 | 18.0% | 5 | 0.0% |
| Asian | 52 | 17.7% | 27 | 13.4% |
| Black or African American | 285 | 32.1% | 139 | 24.6% |
| Native Hawaiian or Other Pacific Islander | 7 | 42.9% | 8 | 17.0% |
| White | 617 | 33.5% | 306 | 22.3% |
| Other | 44 | 31.9% | 21 | 16.3% |
N = Number of subjects in each treatment group
n = Number of subjects in each sub-group category by treatment group
One multicentre, single-arm, open-label long-term safety trial (Trial 3) included a total of 517 subjects 2 to 72 years of age (59.6% of subjects were 2-11 years old, 28.2% of subjects were 12-17 years old, 11.8% of subjects were 18-64 years old, and 0.4% of subjects were 65 years of age or older) with a 5% to 95% treatable BSA. Subjects at participating investigator sites (a subset of sites that participated in Trials 1 and 2) who completed Trials 1 or 2 without safety events that precluded further treatment with Staquis were eligible.
Subjects participated in the study in 28-day treatment courses for up to 48 weeks. Subjects received Staquis for a variable number of treatment courses intermittently based on disease severity as determined by the ISGA at the beginning of each 28-day treatment course: subjects received open-label treatment with Staquis twice daily (on-treatment when ISGA was Mild or worse [≥2]) or no treatment (off-treatment when the ISGA was Clear 0 or Almost Clear 1). Discontinuation from the study was to occur if there was no improvement in the subject’s ISGA after 3 consecutive treatment courses of treatment with Staquis.
Trial 3 did not include an efficacy endpoint; Staquis efficacy response based on ISGA determined the extent of intermittent use of Staquis for up to 48 weeks. Overall, subjects received a mean of 6.2 on-treatment courses (out of a possible 13 on-treatment courses including the 28-day treatment period in Trials 1 or 2). The mean number of consecutive on-treatment courses was 3.6 and the mean number of consecutive off-treatment courses was 2.5.
QT study results
Results from a thorough QT study of Staquis applied to 60% BSA in healthy volunteers did not demonstrate QT prolongation. Although healthy volunteers had lower crisaborole concentrations compared to patients with atopic dermatitis, clinical studies of Staquis did not identify any cardiac effects including prolongation of QT interval.
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with Staquis in one or more subsets of the paediatric population for the treatment of atopic dermatitis (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Absorption
The pharmacokinetics (PK) of Staquis were investigated in 33 paediatric subjects 2 to 17 years of age with mild to moderate atopic dermatitis and a mean ± SD BSA involvement of 49 ± 20% (range 27% to 92%). In this study, subjects applied approximately 3 mg/cm² of Staquis ointment (dose range was approximately 6 g to 30 g per application) twice daily for 8 days. Plasma concentrations were quantifiable in all subjects. The mean ± SD maximum plasma concentration (Cmax) and area under the concentration time curve from 0 to 12 hours post dose (AUC0-12) for crisaborole on Day 8 were 127 ± 196 ng/mL and 949 ± 1240 ng*h/mL, respectively. Systemic concentrations of crisaborole were at steady state by Day 8. Based on the ratios of AUC0-12 between Day 8 and Day 1, the mean accumulation factor for crisaborole was 1.9. Systemic exposure (Cmax and AUC0-12) of crisaborole and its main metabolites increased with increasing % BSA treated.
The studies were performed with a different formulation of crisaborole which, unlike Staquis, contained butylhydroxytoluene (BHT). In vitro permeation testing (IVPT) was performed in intact skin to support therapeutic equivalence between the BHT-containing and the no-added BHT formulations. Although the results were inconclusive and highly variable, a possible slight increase in permeation is not expected to influence the benefit-risk profile of the product in patients with up to 40% BSA affected to a clinically relevant extent.
Distribution
Based on an in vitro study, crisaborole is 97% bound to human plasma proteins.
Biotransformation and elimination
Crisaborole is substantially metabolised into inactive metabolites. The main metabolite 5-(4-cyanophenoxy)-2-hydroxyl benzylalcohol (metabolite 1), is formed via multiple CYP enzymes including CYP3A4, 1A2 and hydrolysis; this metabolite is further metabolised into downstream metabolites, among which 5-(4-cyanophenoxy)-2-hydroxyl benzoic acid (metabolite 2), formed via oxidation, is also a main metabolite. PK of metabolites 1 and 2 were assessed in the PK study described above and the systemic concentrations were at or near steady state by Day 8. Based on the ratios of AUC0-12 between Day 8 and Day 1, the mean accumulation factors for metabolites 1 and 2 were 1.7 and 6.3, respectively. The mean ± SD Cmax and AUC0-12 for metabolite 2 on Day 8 were 1850 ± 1830 ng/mL and 18200 ± 18100 ng*h/mL, respectively. Renal excretion of metabolites is the major route of elimination. Approximately 25% of the radiolabelled dose was absorbed and predominantly excreted in the urine.
Drug interactions
Potential for crisaborole to influence the PK of other medicinal products
In vitro studies using human liver microsomes indicated that under the conditions of clinical use, crisaborole and metabolite 1 are not expected to inhibit CYP1A2, 2B6, 2C8, 2C9, 2C19, and 3A4.
In vitro human liver microsomes studies for metabolite 2 showed that it did not inhibit activities of CYP2C19, 2D6, and 3A4; was a weak inhibitor of CYP1A2 and 2B6; and a moderate inhibitor of CYP2C8 and 2C9. The most sensitive enzyme, CYP2C9, was further investigated in a clinical trial using warfarin as a CYP2C9 substrate. The results of this study showed no drug interaction potential.
In vitro studies indicate that under the condition of clinical use, crisaborole and metabolites 1 and 2 are not expected to induce CYP enzymes.
Based on in vitro data, crisaborole is metabolised to some extent (<30%) via CYP3A4 and CYP1A2. Concomitant administration of Staquis and potent CYP3A4 or CYP1A2 inhibitors may result in increases in crisaborole systemic exposure.
In vitro studies showed that crisaborole and metabolite 1 did not inhibit the activities of uridine diphosphate (UDP)-glucuronosyltransferase (UGT) 1A1, 1A4, 1A6, 1A9, 2B7, and 2B15. Metabolite 2 did not inhibit UGT1A4, 1A6, 2B7, and 2B15. Metabolite 2 showed weak inhibition of UGT1A1; however, no clinically significant drug interactions are expected between crisaborole (and its metabolites) and UGT1A1 substrates at therapeutic concentrations. Metabolite 2 showed moderate inhibition of UGT1A9 and may result in a moderate increase of the concentrations of sensitive UGT1A9 substrates, such as propofol. A clinically relevant interaction between metabolite 2 and propofol is not anticipated due to the posology and method of administration of propofol (intravenous infusion or injection with titration to clinical effect for anaesthesia or sedation). Drug interaction studies with sensitive UGT1A9 substrates have not been conducted.
In vitro studies indicate that under the condition of clinical use, crisaborole and metabolites 1 and 2 are not expected to cause clinically significant interactions with substrates of transporters such as P-glycoprotein, breast cancer resistance protein (BCRP) and organic anionic or cationic transporters.
5.3. Preclinical safety data
Preclinical data from studies conducted in vitro or in vivo by the oral and dermal routes of administration reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated-dose toxicity, genotoxicity, juvenile toxicity, or toxicity to reproduction and development.
A drug-related increased incidence of benign granular cell tumours in the uterus with cervix and vagina (combined) was noted in crisaborole-treated female rats at oral doses approximately 2 times the mean human systemic exposure in maximum use conditions. The clinical relevance of this finding is unknown, however given the tumour type and benign status in a single species and single sex, the relevance to humans is considered to be low.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.