STIVARGA Film-coated tablet Ref.[8927] Active ingredients: Regorafenib
Source: European Medicines Agency (EU) Revision Year: 2019 Publisher: Bayer AG, 51368 Leverkusen, Germany
Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic agents, protein kinase inhibitor;
ATC Code: L01XE21
Mechanism of action and pharmacodynamic effects
Regorafenib is an oral tumour deactivation agent that potently blocks multiple protein kinases, including kinases involved in tumour angiogenesis (VEGFR1, -2, -3, TIE2), oncogenesis (KIT, RET, RAF-1, BRAF, BRAFV600E), metastasis (VEGFR3, PDGFR, FGFR) and tumour immunity (CSF1R). In particular, regorafenib inhibits mutated KIT, a major oncogenic driver in gastrointestinal stromal tumours, and thereby blocks tumour cell proliferation. In preclinical studies regorafenib has demonstrated potent antitumour activity in a broad spectrum of tumour models including colorectal, gastrointestinal stromal and hepatocellular tumour models which is likely mediated by its anti-angiogenic and anti-proliferative effects. In addition, regorafenib reduced the levels of tumour associated macrophages and has shown anti-metastatic effects in vivo. Major human metabolites (M-2 and M-5) exhibited similar efficacies, compared to regorafenib in in vitro and in vivo models.
Clinical efficacy and safety
Metastatic colorectal cancer (CRC)
The clinical efficacy and safety of Stivarga have been evaluated in an international, multi-centre, randomised, double-blind, placebo-controlled phase III study (CORRECT) in patients with metastatic colorectal cancer who have progressed after failure of standard therapy.
The primary efficacy endpoint was Overall Survival (OS). Secondary endpoints were Progression-Free Survival (PFS), Objective Tumour Response Rate (ORR) and Disease Control Rate (DCR).
In total, 760 patients were randomised 2:1 to receive 160 mg regorafenib (4 tablets Stivarga each containing 40 mg regorafenib) orally once daily (N=505) plus Best Supportive Care (BSC or matching placebo (N=255) plus BSC for 3 weeks on therapy followed by 1 week off therapy. The mean daily regorafenib dose received was 147 mg.
Patients continued therapy until disease progression or unacceptable toxicity. A pre-planned interim analysis for efficacy was performed when 432 deaths had occurred. The study was un-blinded after this planned interim analysis of OS had crossed the pre-specified efficacy boundary.
Of the 760 randomised patients, the median age was 61 years, 61% were male, 78% were Caucasian, and all patients had baseline ECOG Performance Status (PS of 0 or 1. PS ≥2 was reported during Stivarga treatment in 11.4% of patients. The median treatment duration and daily dose, as well as the rate of dose modification and dose reduction were similar to those observed in patients with a reported PS ≥2 receiving placebo (8.3%). The majority of patients with PS ≥2 discontinued treatment for progressive disease. The primary site of disease was colon (65%), rectum (29%), or both (6%). A KRAS mutation was reported in 57% of patients at study entry.
Most patients (52%) received 3 or fewer previous lines of treatment for metastatic disease. Therapies included treatment with fluoropyrimidine-based chemotherapy, an anti-VEGF therapy, and, if the patient was KRAS wild type, an anti-EGFR therapy.
The addition of Stivarga to BSC resulted in significantly longer survival, compared to placebo plus BSC with a p value of 0.005178 from stratified log rank test, a hazard ratio of 0.774 [95% CI 0.636, 0.942] ) and a median OS of 6.4 months vs. 5.0 months (see Table 5 and Figure 1). PFS was significantly longer in patients receiving Stivarga plus BSC (hazard ratio: 0.494, p<0.000001, see Table 5). The response rate (complete response or partial response) was 1% and 0.4% for Stivarga and placebo treated patients, respectively (p=0.188432, 1-sided). The DCR (complete response or partial response or stable disease) was significantly higher in patients treated with Stivarga (41.0% vs. 14.9%, p<0.000001, 1 sided).
Table 5. Efficacy results from the CORRECT study:
| Efficacy parameter | Hazard ratio* (95% CI) | P-value (one-sided) | Median (95% CI) | |
|---|---|---|---|---|
| Stivarga plus BSC§ (N=505) | Placebo plus BSC§ (N=255) | |||
| OS | 0,774 (0,636-0,942) | 0,005178 | 6,4 months (5,9-7,3) | 5,0 months (4,4-5,8) |
| PFS** | 0,494 (0,419-0,582) | <0,000001 | 1,9 months (1,9-2,1) | 1,7 months (1,7-1,7) |
§ BSC Supportive Care
* Hazard ratio <1 favours Stivarga
** based on investigator’s assessment of tumour response
Figure 1. Kaplan-Meier curve of OS:
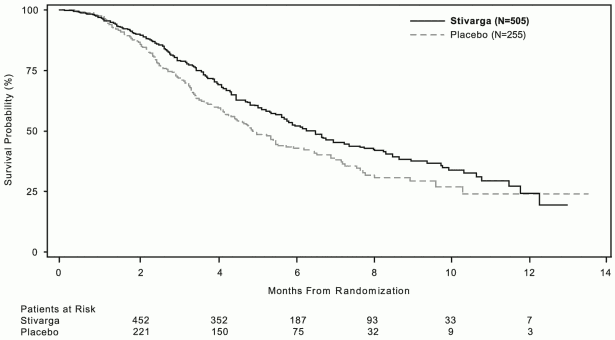
Subgroup analyses for OS and PFS according to age (<65; ≥65), gender, ECOG PS, primary site of disease, time from first diagnosis of metastatic disease, prior anticancer treatment, prior treatment lines for metastatic disease, and KRAS mutation status showed a treatment effect favouring the regorafenib regimen over the placebo regimen. Subgroup analysis results by historical KRAS mutational status showed a treatment effect for OS in favour of regorafenib over placebo for patients with KRAS wild-type tumours whereas a numerically lower effect was reported in patients with KRAS mutant tumours; the treatment effect for PFS favouring regorafenib was observed regardless of KRAS mutational status. The hazard ratio (95% CI) of OS was 0.653 (0.476 to 0.895) for patients with KRAS wild-type tumours and 0.867 (0.670 to 1.123) for patients with KRAS mutant tumours, with no evidence of heterogeneity in treatment effect (non-significant interaction test). The hazard ratio (95% CI) of PFS was 0.475 (0.362 to 0.623) for patients with KRAS wild-type tumours and 0.525 (0.425 to 0.649) for patients with KRAS mutant tumours.
A second phase III, international, multi-centre, randomised, double blind, placebo-controlled study (CONCUR) evaluated the efficacy and safety of Stivarga in 204 pre-treated Asian patients (>90% East Asian) with metastatic colorectal cancer who have progressed after failure of fluoropyrimidinebased chemotherapy. Only 59.5 % of patients enrolled in the CONCUR study were also previously treated with VEGF- or EGFR-targeted agents. The primary efficacy endpoint was OS. The addition of Stivarga to BSC resulted in a significantly longer survival, compared to placebo plus BSC with a hazard ratio of 0.550 (p = 0.000159 stratified log rank test) and a median OS of 8.8 months vs. 6.3 months [95% CI 0.395, 0.765]. PFS was also significantly longer in patients receiving Stivarga plus BSC (hazard ratio: 0.311, p<0.000001), median PFS 3.2 months with Stivarga vs. 1.7 months with placebo. The safety profile of Stivarga plus BSC in the CONCUR study was consistent with the safety profile observed in the CORRECT study.
Gastrointestinal stromal tumours (GIST)
The clinical efficacy and safety of Stivarga have been evaluated in an international, multi-centre, randomised, double-blind, placebo-controlled phase III study (GRID) in patients with gastrointestinal stromal tumours (GIST) previously treated with 2 tyrosine kinase inhibitors (imatinib and sunitinib).
The analysis of the primary efficacy endpoint Progression-Free Survival (PFS) was conducted after 144 PFS events (central blinded assessment). Secondary endpoints including Time To Progression (TTP) and Overall Survival (OS (interim analysis) were also assessed.
In total, 199 patients with GIST were randomised 2:1 to receive either 160 mg regorafenib plus Best Supportive Care (BSC) (N=133) orally once daily or matching placebo plus BSC (N=66) for 3 weeks on therapy followed by 1 week off therapy. The mean daily regorafenib dose received was 140 mg.
Patients continued therapy until disease progression or unacceptable toxicity. Patients receiving placebo who experienced disease progression were offered open-label regorafenib (cross-over option). Patients receiving regorafenib who experienced disease progression and for whom in the investigator's opinion, treatment with regorafenib was providing clinical benefit, were offered the opportunity to continue open-label regorafenib.
Of the 199 randomised patients, the mean age was 58 years, 64% were male, 68% were Caucasian, and all patients had baseline ECOG Performance Status (PS of 0 or 1. The overall median time since most recent progression or relapse to randomisation was 6 weeks.
Regorafenib plus BSC resulted in significantly longer PFS, compared to placebo plus BSC with a hazard ratio of 0.268 [95% CI 0.185, 0.388] and a median PFS of 4.8 months vs. 0.9 months (p < 0.000001). The relative risk of disease progression or death was reduced by approximately 73.2% in regorafenib-treated patients, compared to placebo treated patients (see Table 6, Figure 2).The increase in PFS was consistent independent of age, sex, geographic region, prior lines of treatment, ECOG PS.
TTP was significantly longer in patients receiving regorafenib plus BSC than in patients receiving placebo plus BSC with a hazard ratio of 0.248 [95% CI 0.170, 0.364], and median TTP of 5.4 months vs. 0.9 months (p<0.000001) (see Table 6).
The HR for OS was 0.772 (95% CI, 0.423, 1.408; p = 0.199; median OS not reached in either arm); 85% of patients initially randomised to the placebo arm received post-progression treatment with regorafenib (see Table 6, Figure 3).
Table 6. Efficacy results from the GRID study:
| Efficacy parameter | Hazard ratio* (95% CI) | P-value (one-sided) | Median (95% CI) | |
|---|---|---|---|---|
| Stivarga plus BSC§ (N=133) | Placebo plus BSC§ (N=66) | |||
| PFS | 0,268 (0,185-0,388) | <0,000001 | 4,8 μήνες (4,0-5,7) | 0,9 μήνες (0,9-1,1) |
| TTP | 0,248 (0,170-0,364) | <0,000001 | 5,4 μήνες (4,1-5,7) | 0,9 μήνες (0,9-1,1) |
| OS | 0,772 (0,423-1,408) | 0,199 | NR** | NR** |
§ Best Supportive Care
* Hazard ratio <1 favours Stivarga
** NR: not reached
Figure 2. Kaplan-Meier curves of PFS:
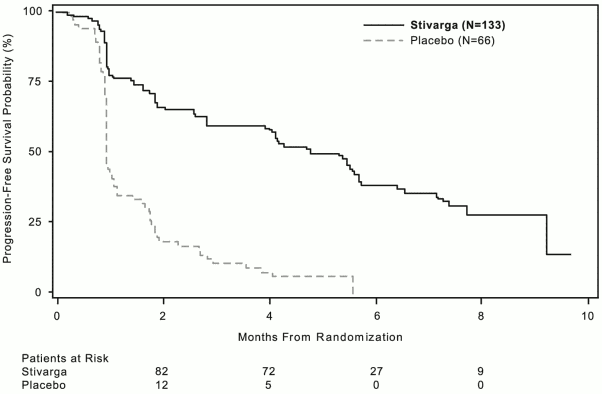
Figure 3. Kaplan-Meier curves of OS:
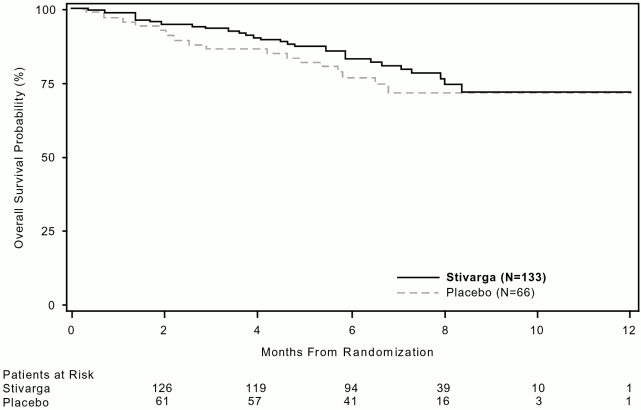
In addition, 56 placebo plus BSC patients received open-label Stivarga after cross-over following disease progression and a total of 41 Stivarga plus BSC patients continued Stivarga treatment after disease progression. The median secondary PFS (as measured by the investigator's assessment) were 5.0 and 4.5 months, respectively.
Hepatocellular carcinoma (HCC)
The clinical efficacy and safety of Stivarga have been evaluated in an international, multi-centre, randomised, double-blind, placebo-controlled phase III study (RESORCE) in patients with hepatocellular carcinoma who have been previously treated with sorafenib.
The primary efficacy endpoint was Overall Survival (OS). Secondary endpoints were Progression-Free Survival (PFS), Time To Progression (TTP), Objective Tumour Response Rate (ORR) and Disease Control Rate (DCR).
In total, 573 patients with HCC were randomised 2:1 to receive either 160 mg regorafenib orally once daily (n=379) plus Best Supportive Care (BSC) or matching placebo (n=194) plus BSC for 3 weeks on therapy followed by 1 week off therapy. The mean daily regorafenib dose received was 144 mg. Patients were eligible to participate in the study if they experienced radiological disease progression during treatment with sorafenib and if they had a liver function status of Child-Pugh class A. Patients who permanently discontinued sorafenib therapy due to sorafenib-related toxicity or who tolerated less than 400 mg sorafenib once daily prior to withdrawal were excluded from the study. Randomisation was performed within 10 weeks after the last treatment with sorafenib. Patients continued therapy with Stivarga until clinical or radiological disease progression or unacceptable toxicity. However, patients could continue Stivarga therapy past progression at the discretion of the investigator.
Demographics and baseline disease characteristics were comparable between the Stivarga- and placebo-treated groups and are shown below for all 573 randomised patients:
- Median age: 63 years
- Male: 88%
- Caucasian: 36%, Asian: 41%
- ECOG Performance Status (PS) of 0: 66% or ECOG PS of 1: 34%
- Child-Pugh A: 98%, Child-Pugh B: 2%
- Aetiology included Hepatitis B (38%), Hepatitis C (21%), Non-Alcoholic Steato Hepatitis
(NASH, 7%)
* Absence of both macroscopic vascular invasion and extra-hepatic tumour spread: 19%
* Barcelona Clinic Liver Cancer (BCLC) stage B: 13%; BCLC stage C: 87%
* Loco-regional transarterial embolisation or chemoinfusion procedures: 61%
* Radiotherapy prior to regorafenib treatment: 15%
* Median duration of sorafenib treatment: 7.8 months
The addition of Stivarga to BSC resulted in a statistically significant improvement in OS compared to placebo plus BSC with a hazard ratio of 0.624 [95% CI 0.498, 0.782], p=0.000017 stratified log rank test, and a median OS of 10.6 months vs. 7.8 months (see Table 7 and Figure 4).
Table 7: Efficacy results from the RESORCE study
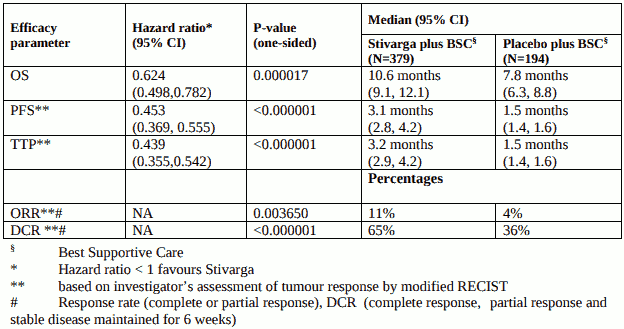
Figure 4: Kaplan-Meier curve of OS
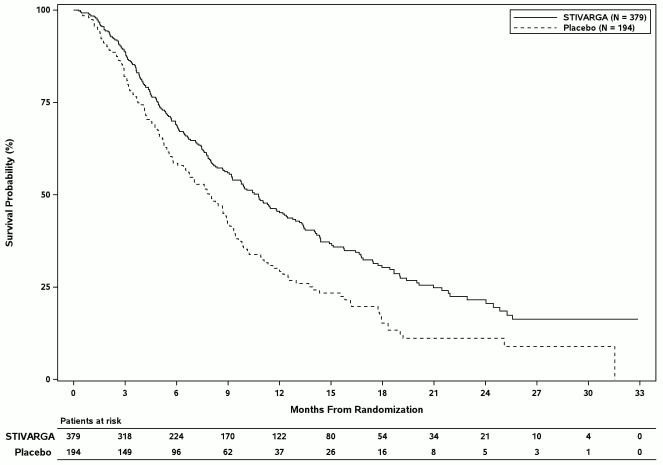
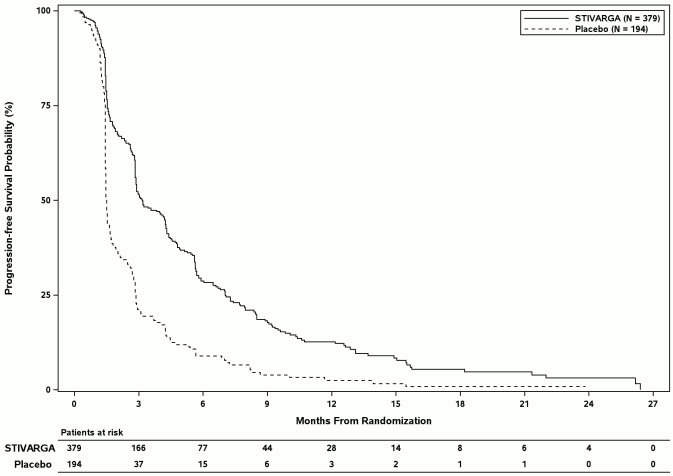
Figure 5: Kaplan-Meier curve of PFS (mRECIST)
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with Stivarga in all subsets of the paediatric population in the treatment of adenocarcinoma of the colon and rectum (see section 4.2 for information on paediatric use). The European Medicines Agency has deferred the obligation to submit the results of studies with Stivarga in one or more subsets of the paediatric population in the treatment of solid malignant tumours (see section 4.2 for information on paediatric use). The European Medicines Agency has waived the obligation to submit the results of studies with Stivarga in all subsets of the paediatric population in the treatment of hepatocellular carcinoma (see section 4.2 for information on paediatric use).
Pharmacokinetic properties
Absorption
Regorafenib reaches mean peak plasma levels of about 2.5 mg/l at about 3 to 4 hours after a single oral dose of 160 mg given as 4 tablets each containing 40 mg. Following single doses of 60 mg or 100 mg, the average relative bioavailability of tablets compared to an oral solution was 69% and 83%, respectively.
The concentrations of regorafenib and its major pharmacologically active metabolites (M-2 and M-5) were highest when given after a low-fat (light) breakfast, compared to either a high-fat breakfast or fasting condition. The exposure for regorafenib was increased by 48% when administered with a high-fat breakfast, and 36% when administered with a low fat breakfast, compared to fasting. The exposure of metabolites M-2 (N-oxide) and M-5 (N-oxide and N-desmethyl) is higher when regorafenib is given with a low fat breakfast, compared to fasting condition and lower when given with a high fat meal, compared to fasting condition.
Distribution
Plasma concentration-time profiles for regorafenib as well as for the major circulating metabolites showed multiple peaks across the 24-hour dosing interval, which are attributed to enterohepatic circulation. In vitro protein binding of regorafenib to human plasma proteins is high (99.5%). In vitro protein binding of M-2 and M-5 is higher (99.8% and 99.95%, respectively) than that of regorafenib. Metabolites M-2 and M-5 are weak substrates of P-gp. Metabolite M-5 is a weak BCRP-substrate.
Biotransformation
Regorafenib is metabolized primarily in the liver by oxidative metabolism mediated by CYP3A4, as well as by glucuronidation mediated by UGT1A9. Two major and six minor metabolites of regorafenib have been identified in plasma. The main circulating metabolites of regorafenib in human plasma are M-2 (N-oxide) and M-5 (N-oxide and N-desmethyl), which are pharmacologically active and have similar concentrations as regorafenib at steady state. M-2 is further metabolised by oxidative metabolism mediated by CYP3A4, as well as by glucuronidation mediated by UGT1A9. Metabolites may be reduced or hydrolysed in the gastrointestinal tract by microbial flora, allowing reabsorption of the unconjugated active substance and metabolites (enterohepatic circulation).
Elimination
Following oral administration, mean elimination half-life for regorafenib and its metabolite M-2 in plasma ranges from 20 to 30 hours in different studies. The mean elimination half-life for the metabolite M-5 is approximately 60 hours (range from 40 to 100 hours).
Approximately 90% of the radioactive dose was recovered within 12 days after administration, with about 71% of the dose excreted in faeces (47% as parent compound, 24% as metabolites), and about 19% of the dose excreted in urine as glucuronides. Urinary excretion of glucuronides decreased below 10% under steady-state conditions. Parent compound found in faeces could be derived from intestinal degradation of glucuronides or reduction of metabolite M-2 (N-oxide), as well as unabsorbed regorafenib. M-5 may be reduced to M-4 in the gastrointestinal tract by microbial flora, allowing reabsorption of M-4 (enterohepatic circulation). M-5 is finally excreted via M-4 as M-6 (carboxylic acid) in faeces.
Linearity / non-linearity
Systemic exposure of regorafenib at steady-state increases dose proportionally up to 60 mg and less than proportionally at doses greater than 60 mg. Accumulation of regorafenib at steady state results in about a 2-fold increase in plasma concentrations, which is consistent with the elimination half-life and dosing frequency. At steady state, regorafenib reaches mean peak plasma levels of about 3.9 mg/L (8.1 micromolar) after oral administration of 160 mg regorafenib and the peak-to-trough ratio of mean plasma concentrations is less than 2. Both metabolites, M-2 and M-5, exhibit non-linear accumulation, which might be caused by enterohepatic recycling or saturation of the UGT1A9 pathway. Whereas plasma concentrations of M-2 and M-5 after a single dose of regorafenib are much lower than those of parent compound, steady-state plasma concentrations of M-2 and M-5 are comparable to those of regorafenib.
Hepatic impairment
The exposure of regorafenib and its metabolites M-2 and M-5 is comparable in patients with mild hepatic impairment (Child-Pugh A) and patients with normal hepatic function. Limited data in patients with moderate hepatic impairment (Child-Pugh B) indicate similar exposure, compared to patients with normal hepatic function after a single 100 mg dose of regorafenib. There are no data for patients with Child-Pugh C (severe) hepatic impairment. Regorafenib is mainly eliminated via the liver, and exposure might be increased in this patient population.
Renal impairment
Available clinical data and physiology-based pharmacokinetic modelling indicate similar steady-state exposure of regorafenib and its metabolites M-2 and M-5 in patients with mild or moderate renal impairment, compared to patients with normal renal function. In patients with severe renal impairment compared to patients with normal renal function, regorafenib exposure was similar while exposure to M-2 and M-5 was decreased by about 30% under steady-state conditions, which is not considered clinically relevant.
The pharmacokinetics of regorafenib has not been studied in patients with end-stage renal disease. However, physiology-based pharmacokinetic modelling does not predict any relevant change in exposure in these patients.
Elderly
Age did not affect the regorafenib pharmacokinetics over the studied age range (29-85 years).
Gender
The pharmacokinetics of regorafenib is not influenced by gender.
Ethnic differences
The exposure of regorafenib in various Asian populations (Chinese, Japanese, Korean) is within the same range as seen in Caucasians.
Cardiac electrophysiology / QT prolongation
No QTc prolonging effects were observed after administration of 160 mg regorafenib at steady state in a dedicated QT study in male and female cancer patients.
Preclinical safety data
Systemic toxicity
After repeated dosing to mice, rats and dogs, adverse effects were observed in a number of organs, primarily in the kidneys, liver, digestive tract, thyroid gland, lympho-/haematopoietic system, endocrine system, reproductive system and skin. A slightly increased incidence of thickening of the atrioventricular valves of the heart was seen in the 26 week repeat-dose toxicity study in rats. This may be due to acceleration of an age-related physiological process. These effects occurred at systemic exposures in the range of or below the anticipated human exposure (based on AUC comparison).
Alterations of teeth and bones and adverse effects in the reproductive system were more pronounced in young and growing animals as well as in juvenile rats and indicate a potential risk for children and adolescents.
Reproductive and developmental toxicity
Specific studies on fertility have not been performed. However, a potential of regorafenib to adversely affect male and female reproduction has to be considered based on morphological changes in the testes, ovaries, and the uterus observed after repeated dosing in rats and dogs at exposures below the anticipated human exposure (based on AUC comparison). The observed changes were only partially reversible.
An effect of regorafenib on intrauterine development was shown in rabbits at exposures below the anticipated human exposure (based on AUC comparison). Main findings consisted of malformations of the urinary system, the heart and major vessels, and the skeleton.
Genotoxicity and carcinogenicity
There was no indication for a genotoxic potential of regorafenib tested in standard assays in vitro and in vivo in mice.
Studies on the carcinogenic potential of regorafenib have not been performed.
Environmental risk assessment (ERA)
Environmental risk assessment studies have shown that regorafenib has the potential to be persistent, bioaccumulative and toxic to the environment and may pose a risk to the surface water and to the sediment compartment (see section 6.6).
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.