SUNOSI Film-coated tablet Ref.[10411] Active ingredients: Solriamfetol
Source: FDA, National Drug Code (US) Revision Year: 2020
12.1. Mechanism of Action
The mechanism of action of solriamfetol to improve wakefulness in patients with excessive daytime sleepiness associated with narcolepsy or obstructive sleep apnea is unclear. However, its efficacy could be mediated through its activity as a dopamine and norepinephrine reuptake inhibitor (DNRI).
12.2. Pharmacodynamics
Solriamfetol binds to the dopamine transporter and norepinephrine transporter with low affinity (Ki=14.2 µM and 3.7 µM, respectively), and inhibits the reuptake of dopamine and norepinephrine with low potency (IC50 =2.9 μM and 4.4 μM, respectively). Solriamfetol has no appreciable binding affinity for the serotonin transporter (Ki=81.5 µM) and does not inhibit serotonin reuptake (IC50 >100 μM). Solriamfetol has no appreciable binding affinity to dopamine, serotonin, norepinephrine, GABA, adenosine, histamine, orexin, benzodiazepine, muscarinic acetylcholine, or nicotinic acetylcholine receptors.
Cardiac Electrophysiology
The effect of solriamfetol 300 mg and 900 mg (twice and six times the maximum recommended dose, respectively) on the QTc interval was evaluated in a randomized, double-blind, placebo-, and positive-controlled (moxifloxacin 400 mg), 4-period, crossover study in 60 healthy subjects. A large increase in heart rate was observed in both solriamfetol treatment groups (mean change from baseline in HR of 21 and 27 bpm in the 300 and 900 mg groups, respectively, compared with 8 bpm in the placebo group). These heart rate effects impact the interpretability of the QTc effects, particularly in the 900 mg group. In this study, solriamfetol 300 mg did not prolong the QTcF interval to a clinically relevant extent.
12.3. Pharmacokinetics
Solriamfetol exhibits linear kinetics over the dose range of 42 to 1008 mg (approximately 0.28 to 6.7 times the maximum recommended dosage). Steady state is reached in 3 days, and once‑daily administration is expected to result in minimal accumulation (1.06 times single‑dose exposure).
Absorption
The oral bioavailability of solriamfetol is approximately 95%. Peak plasma concentration of solriamfetol occurs at a median Tmax of 2 hours (range 1.25 to 3.0 hours) post-dose under fasted conditions.
Effect of Food
Ingestion of solriamfetol with a high-fat meal resulted in minimal change in Cmax and AUCinf; however, a delay of approximately 1 hour in Tmax was observed.
Distribution
The apparent volume of distribution of solriamfetol is approximately 199 L. Plasma protein binding ranged from 13.3% to 19.4% over solriamfetol concentration range of 0.059 to 10.1 mcg/mL in human plasma. The mean blood‑to‑plasma concentration ratio ranged from 1.16 to 1.29.
Elimination
Solriamfetol exhibits first‑order elimination after oral administration. The apparent mean elimination half‑life is about 7.1 hours.
Metabolism
Solriamfetol is minimally metabolized in humans.
Excretion
Approximately 95% of the dose was recovered in urine as unchanged solriamfetol, and 1% or less of the dose was recovered as the minor inactive metabolite N‑acetyl solriamfetol in a mass balance study. Renal clearance (18.2 L/h) represented the majority of apparent total clearance (19.5 L/h). Active tubular secretion is likely involved in the renal elimination of the parent drug.
Specific Populations
Population PK analysis indicated that age, gender, and race do not have clinically relevant effects on the pharmacokinetics of solriamfetol. No dose adjustments were made in clinical studies that enrolled patients ages 65 and above.
Patients with Renal Impairment
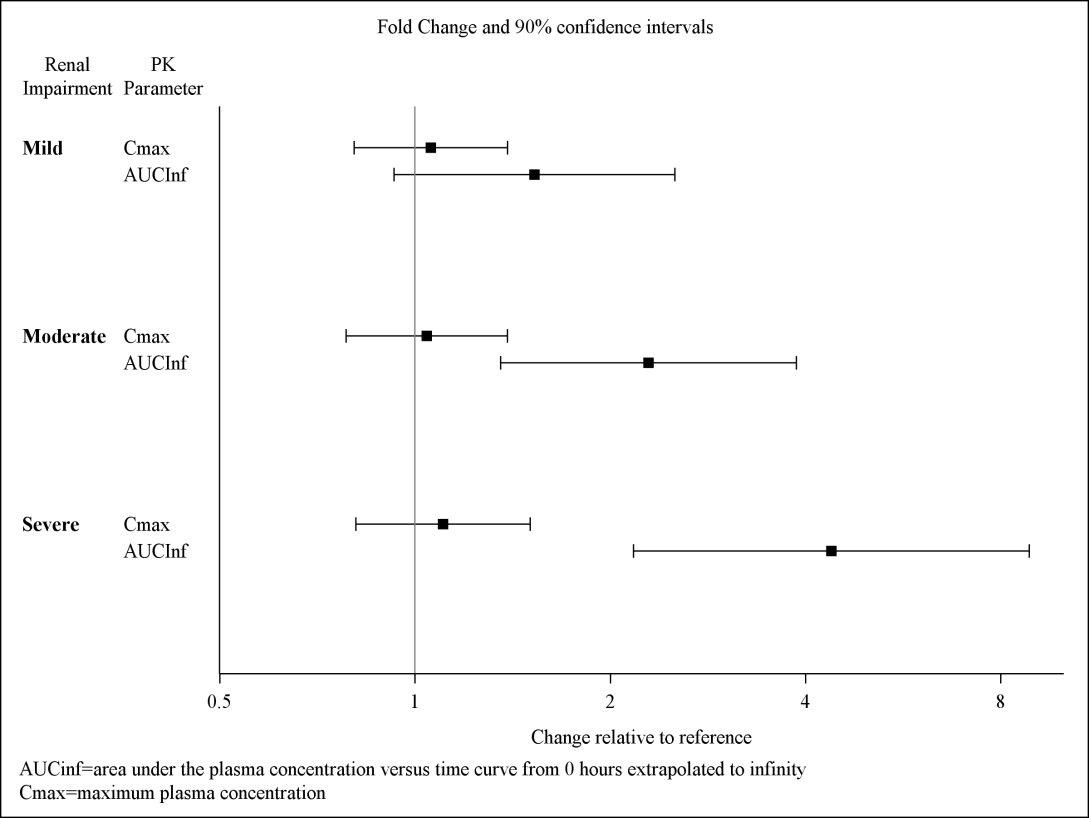
Exposures to solriamfetol in patients with renal impairment compared to subjects with normal renal function (eGFR ≥90 mL/min/1.73 m²) are summarized in Figure 1. The half‑life of solriamfetol was increased approximately 1.2‑, 1.9‑, and 3.9‑fold in patients with mild (eGFR 60‑89 mL/min/1.73 m²), moderate (eGFR 30–59 mL/min/1.73 m²), or severe (eGFR <30 mL/min/1.73 m²) renal impairment, respectively. Exposure (AUC) and half-life of solriamfetol was significantly increased in patients with ESRD (eGFR <15 mL/min/1.73 m²) [see Use in Specific Populations (8.6)]. An average of 21% of solriamfetol was removed by hemodialysis. In general, median Tmax values were not affected by renal impairment.
Figure 1. Effect of Renal Impairment on Solriamfetol Pharmacokinetics:
Drug Interaction Studies
In Vitro Studies
CYP and UGT Enzymes: Solriamfetol was minimally metabolized in vitro. Solriamfetol is not an inhibitor of CYPs 1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, or 3A4. It does not induce CYP1A2, 2B6, 3A4, or UGT1A1 enzymes at clinically relevant concentrations.
Transporter Systems: Solriamfetol is a low-avidity substrate of OCT2, MATE1, OCTN1, and OCTN2. Solriamfetol is a weak inhibitor of OCT2 (IC50 of 146 μM) and MATE1 (IC50 of 211 μM), and is not an inhibitor of OCT1, MATE2-K, OCTN1, or OCTN2. Solriamfetol does not appear to be a substrate or inhibitor of P-gp, BCRP, OATP1B1, OATP1B3, OAT1, or OAT3.
Based on in vitro data, clinically significant PK drug interactions with major CYPs and transporters are not expected in patients taking SUNOSI.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Solriamfetol did not increase the incidence of tumors in rats or mice treated orally for up to 101 and 104 weeks at 35, 80, and 200 mg/kg/day (rat), and 20, 65, and 200 mg/kg/day (mouse), respectively. These doses are approximately 2, 6, and 18 times (rat), and 0.4, 2.6, and 7 times (mouse) the MRHD based on AUC.
Mutagenesis
Solriamfetol was not mutagenic in the in vitro bacterial reverse mutation (Ames) assay or clastogenic in the in vitro mammalian chromosomal aberration assay or in the in vivo mouse bone marrow micronucleus assay.
Impairment of Fertility
Solriamfetol did not affect fertility or sperm parameters when administered orally to male rats for 8 weeks at doses of 35 and 110 mg/kg/day, which are approximately 2 and 7 times the MRHD, based on mg/m² body surface area. At 350 mg/kg/day, which is approximately 22 times the MRHD based on mg/m² body surface area, solriamfetol decreased sperm count and sperm concentration without affecting fertility.
Solriamfetol did not affect fertility when administered orally to female rats for 2 weeks premating, during mating, and through gestation day 7 at 15, 67, and 295 mg/kg/day, which are approximately 1, 4, and 19 times the MRHD, based on mg/m² body surface area.
14. Clinical Studies
14.1 Narcolepsy
The efficacy of SUNOSI in improving wakefulness and reducing excessive daytime sleepiness was demonstrated in a 12‑week, multi‑center, randomized, double‑blind, placebo‑controlled, parallel-group study (Study 1; NCT02348593) in adult patients with a diagnosis of narcolepsy according to the ICSD‑3 or DSM‑5 criteria.
Wakefulness and sleepiness were assessed using the Maintenance of Wakefulness Test (MWT) and the Epworth Sleepiness Scale (ESS). The MWT measures an individual's ability to remain awake during the daytime in a darkened, quiet environment. Patients were instructed to remain awake for as long as possible during 40‑minute test sessions, and sleep latency was determined as the mean number of minutes patients could remain awake in the first four test sessions. The ESS is an 8‑item questionnaire by which patients rate their perceived likelihood of falling asleep during usual daily life activities. Change in overall symptom severity was assessed using the Patient Global Impression of Change (PGIc) scale. The PGIc is a 7‑point patient-reported scale by which patients rate their symptom change since the beginning of the study. Responses range from "very much improved" to "very much worse". The co-primary efficacy endpoints were change from baseline in MWT and ESS at Week 12. A pre-specified secondary endpoint was percentage of subjects reported as improved (minimally, much, or very much) at Week 12 by PGIc.
A total of 239 patients with narcolepsy were randomized to receive SUNOSI 75 mg, 150 mg, or 300 mg (two times the maximum recommended daily dose), or placebo once daily. Patients randomized to the 150-mg dose received 75 mg for the first 3 days before increasing to 150 mg.
Demographic and baseline disease characteristics were similar for the SUNOSI and placebo groups. Median age was 34 years (range 18 to 70 years), 65% were female, 80% were Caucasian, 14% were African American, and 3% were Asian. Approximately 51% of patients had cataplexy.
Compared to the placebo group, patients randomized to 150 mg SUNOSI showed statistically significant improvements on the MWT (treatment effect difference: 7.7 minutes, Table 6) and on the ESS (treatment effect difference: 3.8 points, Table 7) at Week 12. These effects were apparent at Week 1 and consistent with the results at Week 12. The change on percentage of subjects reported as improved by PGIc was also statistically significant compared with placebo. There were trends toward improvement in the SUNOSI 75-mg treatment group (Tables 6 and 7); however, these changes were not statistically significant. There was no evidence of differential efficacy in patients with cataplexy and patients without cataplexy. Examination of subgroups by age, race, and sex did not suggest differences in response.
At Week 12, 150 mg of SUNOSI demonstrated improvements in wakefulness compared to placebo as assessed in test sessions 1 (approximately 1 hour post‑dose) through 5 (approximately 9 hours post‑dose) of the MWT (Figure 2). Nighttime sleep as measured with polysomnography was not affected by the use of SUNOSI in Study 1.
Figure 2. Maintenance of Wakefulness Test Improvements in Test Sessions 1 throug h 5 in Patients with Narcolepsy in Study 1 at Week:

14.2 Obstructive Sleep Apnea (OSA)
The efficacy of SUNOSI in improving wakefulness and reducing excessive daytime sleepiness in patients with OSA was demonstrated in a 12-week multi‑center, randomized, double-blind, placebo‑controlled study (Study 2; NCT02348606) in adults diagnosed with OSA according to ICSD‑3 criteria. The co-primary efficacy endpoints were change from baseline in MWT and ESS at Week 12; a pre-specified secondary endpoint was percentage of subjects reported as improved (minimally, much, or very much) at Week 12 by PGIc.
A total of 476 patients with OSA were randomized to receive SUNOSI 37.5 mg, 75 mg, 150 mg, or 300 mg (two times the maximum recommended daily dose), or placebo once daily. Patients randomized to the 150-mg dose received 75 mg for the first 3 days before increasing to 150 mg.
Demographic and baseline disease characteristics were similar for the SUNOSI and placebo groups. Median age was 55 years (range 20 to 75 years), 37% were female, 76% were Caucasian, 19% were African American, and 4% were Asian.
Compared to the placebo group, patients randomized to 37.5 mg, 75 mg, and 150 mg SUNOSI showed statistically significant improvements on the MWT (treatment effect difference: 4.5 minutes, 8.9 minutes, and 10.7 minutes respectively; Table 6) and ESS (treatment effect difference: 1.9 points, 1.7 points, and 4.5 points respectively; Table 7) at Week 12. These effects were apparent at Week 1 and consistent with the results at Week 12. The change on percentage of subjects reported as improved by PGIc was also statistically significant compared with placebo. Examination of subgroups by age, race, and sex did not suggest differences in response.
At Week 12, 37.5 mg, 75 mg, and 150 mg of SUNOSI all demonstrated improvements in wakefulness compared to placebo as assessed in test sessions 1 (approximately 1 hour post‑dose) through 5 (approximately 9 hours post‑dose) of the MWT (Figure 3). Nighttime sleep as measured with polysomnography was not affected by the use of SUNOSI in Study 2. Patients' compliance with a primary OSA therapy device was similar across the placebo and SUNOSI treatment groups at baseline, and did not change during the 12‑week study period in any treatment group.
Figure 3. Maintenance of Wakefulness Test Improvements in Test Sessions 1 through 5 in Patients with OSA in Study 2 at Week:
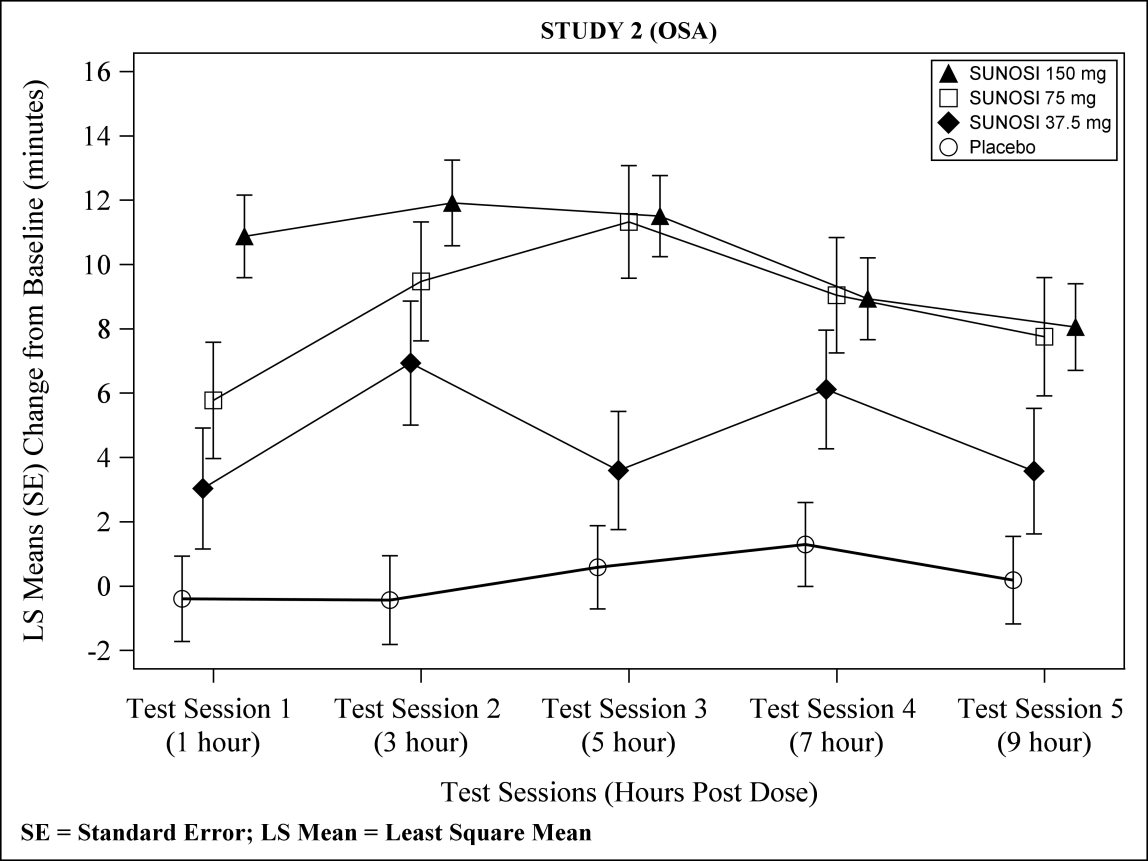
Table 6. Efficacy Results for Maintenance of Wakefulness Test (minutes) in Patients with Narcolepsy (Study 1) and OSA (Study 2):
| Indication/Study | Treatment Group (N) | Baseline Mean (SD) | LS Mean Change from Baseline at Week 12 (SE) | Difference from Placebo (95% CI) |
|---|---|---|---|---|
| Narcolepsy STUDY 1 | Placebo (58) | 6.2 (5.7) | 2.1 (1.3) | - |
| SUNOSI 75 mg (59) | 7.5 (5.4) | 4.7 (1.3) | 2.6 (-1.0, 6.3) | |
| SUNOSI 150 mg* (55) | 7.9 (5.7) | 9.8 (1.3) | 7.7 (4.0, 11.3) | |
| OSA STUDY 2 | Placebo (114) | 12.6 (7.1) | 0.2 (1.0) | - |
| SUNOSI 37.5 mg* (56) | 13.6 (8.1) | 4.7 (1.4) | 4.5 (1.2, 7.9) | |
| SUNOSI 75 mg* (58) | 12.4 (6.9) | 9.1 (1.4) | 8.9 (5.6, 12.4) | |
| SUNOSI 150 mg* (116) | 12.5 (7.2) | 11.0 (1.0) | 10.7 (8.1, 13.4) |
SD = standard deviation; SE = standard error; LS Mean = least square mean; CI = confidence interval
Maximum possible MWT score is 40 minutes. A positive change represents improvement.
Difference from Placebo = LS Mean Difference between change from baseline between active drug and placebo.
* Dose that was statistically significantly superior to placebo after adjusting for multiplicity.
Table 7. Efficacy Results for Epworth Sleepiness Scale in Patients with Narcolepsy (Study 1) and OSA (Study 2):
| Indication/Study | Treatment Groups (N) | Baseline Score Mean (SD) | LS Mean Change from Baseline at Week 12 (SE) | Difference from Placebo (95% CI) |
|---|---|---|---|---|
| Narcolepsy STUDY 1 | Placebo (58) | 17.3 (2.9) | -1.6 (0.7) | - |
| SUNOSI 75 mg (59) | 17.3 (3.5) | -3.8 (0.7) | -2.2 (-4.0, -0.3) | |
| SUNOSI 150 mg* (55) | 17.0 (3.6) | -5.4 (0.7) | -3.8 (-5.6, -2.0) | |
| OSA STUDY 2 | Placebo (114) | 15.6 (3.3) | -3.3 (0.5) | - |
| SUNOSI 37.5 mg* (56) | 15.1 (3.5) | -5.1 (0.6) | -1.9 (-3.4, -0.3) | |
| SUNOSI 75 mg* (58) | 15.0 (3.5) | -5.0 (0.6) | -1.7 (-3.2, -0.2) | |
| SUNOSI 150 mg* (116) | 15.1 (3.4) | -7.7 (0.4) | -4.5 (-5.7, -3.2) |
SD = standard deviation; SE = standard error; LS Mean = least square mean; CI = confidence interval
Scores range from 0 to 24 with higher scores indicating more severe sleepiness. A negative change represents improvement.
Difference from placebo = LS mean difference between change from baseline between SUNOSI and placebo.
* Dose that was statistically significantly superior to placebo after adjusting for multiplicity.
14.3 Maintenance of Efficacy in Narcolepsy and OSA
The maintenance of effect of SUNOSI in improving wakefulness and reducing excessive daytime sleepiness in patients with narcolepsy and OSA was assessed in two randomized‑withdrawal, placebo‑controlled studies, Study 3 (NCT02348619) and Study 4 (NCT02348632).
Study 3 was a 6‑week, multi-center, double-blind, placebo‑controlled, randomized‑withdrawal study in 174 adult patients with a diagnosis of OSA. The co-primary efficacy endpoints were change from the beginning to the end of the randomized withdrawal period in MWT and ESS. During a 2‑week, open-label titration phase, patients were started on SUNOSI 75 mg once daily, and were titrated to the maximum tolerable dose between 75 mg and 300 mg per day (two times the maximum recommended daily dose). Patients were continued on this dose for a 2‑week stable-dose phase. At the end of the stable‑dose phase, 124 patients who reported "much" or "very much" improvement on the PGIc and who showed improvements on the MWT and ESS entered a double-blind withdrawal phase and were randomized 1:1 to either continue SUNOSI at the dose received in the stable‑dose phase or switch to placebo. Compared to patients who remained on SUNOSI, patients randomized to placebo experienced statistically significant worsening of sleepiness as measured by the MWT and ESS (Table 8).
Study 4 was a 52‑week, open-label study in 638 patients with either narcolepsy or OSA who had completed a prior trial. During a 2‑week, open-label titration phase, patients were started on SUNOSI 75 mg once daily, and were titrated to the maximum tolerable dose between 75 mg and 300 mg per day (two times the maximum recommended daily dose). Patients remained on this dose during a subsequent open‑label treatment period of either 38 (for patients previously enrolled in Study 1 or Study 2) or 50 (all others) weeks. A 2‑week randomized‑withdrawal period was incorporated into the study. After 6 months of stable‑dose treatment, 282 patients (79 with narcolepsy; 203 with OSA) entered the randomized‑withdrawal period. Patients were randomized 1:1 to either continue to receive SUNOSI at the dose received in the maintenance phase or to switch to placebo. The primary efficacy endpoint was change from the beginning to the end of the randomized‑withdrawal period in ESS. Compared to patients who remained on SUNOSI, patients randomized to placebo experienced statistically significant worsening of sleepiness as measured by the ESS (Table 8).
Table 8. Efficacy Results from Randomized Withdrawal Studies in Patients with Narcolepsy and OSA in Studies 3 and 4:
| Indication/Study | Endpoint | Treatment Groups (N) | Beginning of Randomized Withdrawal Period (Baseline) Mean (SD) | LS Mean Change from Baseline (SE) | Difference from Placebo (95% CI) |
|---|---|---|---|---|---|
| OSA STUDY 3 | MWT (minutes) | Placebo (62) SUNOSI* (60) | 29.0 (9.9) 31.7 (9.2) | -12.1 (1.3) -1.0 (1.4) | 11.2 (7.8, 14.6) |
| ESS Score | Placebo (62) SUNOSI* (60) | 5.9 (3.8) 6.4 (4.4) | 4.5 (0.7) -0.1 (0.7) | -4.6 (-6.4, -2.8) | |
| OSA and Narcolepsy STUDY 4 | ESS Score | Placebo (141) SUNOSI* (139) | 7.8 (5.0) 7.3 (5.3) | 5.3 (0.4) 1.6 (0.4) | -3.7 (-4.8, -2.7) |
SD = standard deviation; SE = standard error; LS Mean = least square mean; CI = confidence interval
For MWT, maximum possible score is 40 minutes; negative changes indicate worsening.
For ESS, scores range from 0 to 24; positive changes indicate worsening.
* Statistically significantly superior to placebo after adjusting for multiplicity.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.