TAGRISSO Film-coated tablet Ref.[9022] Active ingredients: Osimertinib
Source: European Medicines Agency (EU) Revision Year: 2025 Publisher: AstraZeneca AB, SE-151 85 Södertälje, Sweden
Pharmacodynamic properties
Pharmacotherapeutic group: antineoplastic agents, protein kinase inhibitors
ATC code: L01EB04
Mechanism of action
Osimertinib is a Tyrosine Kinase Inhibitor (TKI). It is an irreversible inhibitor of EGFRs harboring sensitising-mutations (EGFRm) and TKI-resistance mutation T790M.
Pharmacodynamic effects
In vitro studies have demonstrated that osimertinib has high potency and inhibitory activity against EGFR across a range of all clinically relevant EGFR sensitising-mutant and T790M mutant NSCLC cell lines (apparent IC50s from 6 nM to 54 nM against phospho-EGFR). This leads to inhibition of cell growth, while showing significantly less activity against EGFR in wild-type cell lines (apparent IC50s from 480 nM to 1.8 μM against phospho-EGFR). In vivo oral administration of osimertinib lead to tumour shrinkage in both EGFRm and T790M NSCLC xenograft and transgenic mouse lung tumour models.
Cardiac electrophysiology
The QTc interval prolongation potential of TAGRISSO was assessed in 210 patients who received osimertinib 80 mg daily in AURA2. Serial ECGs were collected following a single dose and at steady-state to evaluate the effect of osimertinib on QTc intervals. A pharmacokinetic/pharmacodynamic analysis predicted a drug-related QTc interval prolongation at 80 mg of 14 msec with an upper bound of 16 msec (90% CI).
Clinical efficacy and safety
Adjuvant treatment of EGFR mutation-positive NSCLC, with or without prior adjuvant chemotherapy – ADAURA
The efficacy and safety of TAGRISSO for the adjuvant treatment of patients with EGFR mutation-positive (Ex19del or L858R) NSCLC who have had complete tumour resection with or without prior adjuvant chemotherapy was demonstrated in a randomised, double-blind, placebo-controlled study (ADAURA).
Eligible patients with resectable tumors stage IB – IIIA (according to American Joint Commission on Cancer [AJCC] 7th edition) were required to have EGFR mutations (Ex19del or L858R), identified by the cobas EGFR Mutation Test performed prospectively using biopsy or surgical specimen in a central laboratory.
Patients were randomised 1:1 to receive TAGRISSO (n=339, 80 mg orally once daily) or placebo (n=343) following recovery from surgery and standard adjuvant chemotherapy where given. Patients not receiving adjuvant chemotherapy were randomised within 10 weeks and patients receiving adjuvant chemotherapy within 26 weeks following surgery. Randomisation was stratified by EGFR mutation type (Ex19del or L858R), ethnicity (Asian or non-Asian) and staging based on pathological tumor-node-metastasis (pTNM) (IB or II or IIIA) according to AJCC 7th edition. Treatment was given until disease recurrence, unacceptable toxicity, or for 3 years.
The major efficacy outcome measure was disease-free survival (DFS) by investigator assessment in the stage II-IIIA population. DFS by investigator assessment in the stage IB-IIIA population (overall population) was an additional efficacy outcome measure. Other additional efficacy outcome measures included DFS rate, overall survival (OS), OS rate, and time to deterioration in health-related quality of life (HRQoL) SF-36.
The baseline demographic and disease characteristics of the overall population were: median age 63 years (range 30-86 years), ≥75 years old (11%), female (70%), Asian (64%), never smokers (72%), World Health Organization (WHO) performance status of 0 (64%) or 1 (36%), stage IB (31%), stage II (34%), and IIIA (35%). With regards to EGFR mutation status, 55% were exon 19 deletions and 45% were exon 21 L858R substitution mutations; 9 patients (1%) also had a concurrent de novo T790M mutation. The majority (60%) of patients received adjuvant chemotherapy prior to randomisation (26% IB; 71% IIA; 73% IIB; 80% IIIA). At the time of the DFS analysis, 205 (61%) patients were still on active treatment; of the 73 (11%) patients who had the opportunity to complete the 3-year treatment period, 40 (12%) were in the osimertinib arm and 33 (10%) in the placebo arm.
There were 37 patients who had disease recurrence on TAGRISSO. The most commonly reported sites of recurrence were: lung (19 patients); lymph nodes (10 patients) and central nervous system (CNS) (5 patients). There were 157 patients who had disease recurrence on placebo. The most commonly reported sites were: lung (61 patients); lymph nodes (48 patients) and CNS (34 patients).
ADAURA demonstrated a statistically significant reduction in the risk of disease recurrence or death for patients treated with TAGRISSO compared to patients treated with placebo in the stage II-IIIA population. Similar results were observed in the stage IB-IIIA population.
Efficacy results from ADAURA by investigator assessment are summarised in Table 3.
Table 3. Efficacy results from ADAURA by investigator assessment:
| Stage II-IIIA population | Stage IB-IIIA population | |||
|---|---|---|---|---|
| Efficacy parameter | TAGRISSO (N=233) | Placebo (N=237) | TAGRISSO (N=339) | Placebo (N=343) |
| Disease-free survival | ||||
| Number of events (%) | 26 (11) | 130 (55) | 37 (11) | 159 (46) |
| Recurrent disease (%) | 26 (11) | 129 (54) | 37 (11) | 157 (46) |
| Deaths (%) | 0 | 1 (0.4) | 0 | 2 (0.6) |
| Median DFS, months (95% CI) | NC (38.8, NC) | 19.6 (16.6, 24.5) | NC (NC, NC) | 27.5 (22.0, 35.0) |
| HR (99.06% CI); P-value | 0.17 (0.11, 0.26); <0.0001a | 0.20 (0.14, 0.30); <0.0001b | ||
| DFS rate at 12 months (%) (95% CI) | 97 (94, 99) | 61 (54, 67) | 97 (95, 99) | 69 (63, 73) |
| DFS rate at 24 months (%) (95% CI) | 90 (84, 93) | 44 (37, 51) | 89 (85, 92) | 52 (46, 58) |
| DFS rate at 36 months (%) (95% CI)c,d | 78 (65, 87) | 28 (19, 38) | 79 (69, 86) | 40 (32, 48) |
HR=Hazard Ratio; CI=Confidence Interval; NC=Not Calculable
DFS results based on investigator assessment.
A HR<1 favours TAGRISSO.
Median follow-up time for DFS was 22.1 months for patients receiving TAGRISSO, 14.9 months for patients receiving placebo (stage II-IIIA population) and 16.6 months for patients receiving placebo (stage IB-IIIA population).
DFS results are from the primary analysis (17 January 2020).
a Adjusted for an interim analysis (33% maturity) a p-value <0.0094 was required to achieve statistical significance.
b Adjusted for an interim analysis (29% maturity) a p-value <0.0088 was required to achieve statistical significance.
c The number of patients at risk at 36 months was 18 patients in the TAGRISSO arm and 9 patients in the placebo arm (stage II-IIIA population).
d The number of patients at risk at 36 months was 27 patients in the TAGRISSO arm and 20 patients in the placebo arm (stage IB-IIIA population).
The final analysis of OS (data cut-off [DCO]: 27 January 2023) demonstrated a statistically significant improvement in OS for patients treated with TAGRISSO compared to placebo for both the stage II-IIIA population (100 OS events [21% maturity]; HR=0.49; 95.03% CI: 0.33, 0.73; p-value=0.0004) and the overall population (IB-IIIA; 124 OS events [18% maturity]; HR=0.49; 95.03% CI: 0.34, 0.70; p-value <0.0001). For both populations, the median OS was not reached in either treatment arm and the 95% CIs were not calculable. The median follow-up time for OS in all patients was 59.9 months (stage II-IIIA population) and 60.4 months (stage IB-IIIA population) in the TAGRISSO arm and 56.2 months (stage II-IIIA population) and 59.4 months (stage IB-IIIA population) in the placebo arm.
Figure 1. Kaplan-Meier curve of disease-free survival in stage II-IIIA patients by investigator assessment:
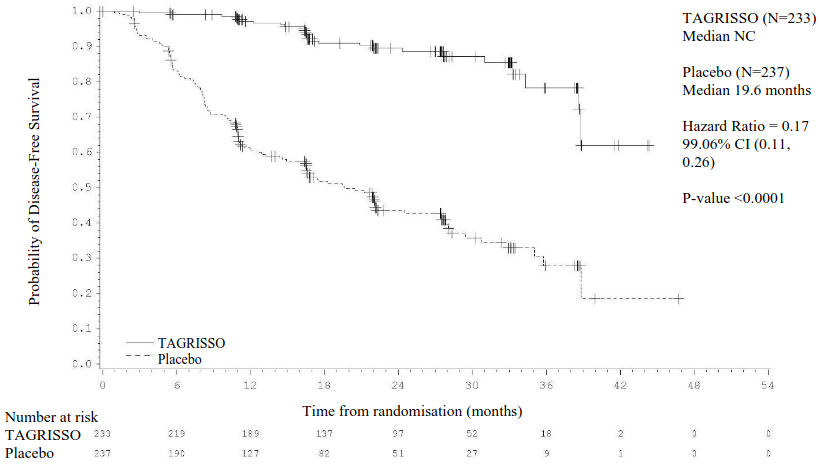
+ Censored patients.
The values at the base of the figure indicate number of subjects at risk.
NC = Not Calculable.
Figure 2. Kaplan-Meier curve of disease-free survival in stage IB-IIIA (overall population) patients by investigator assessment:
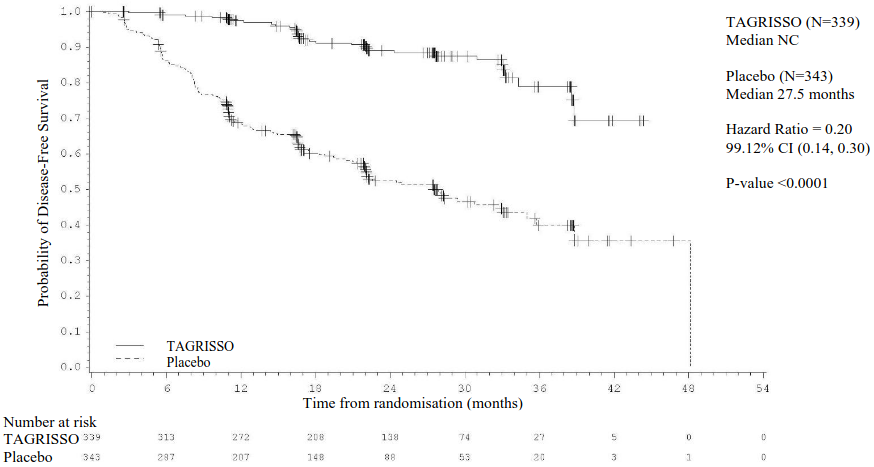
+ Censored patients.
The values at the base of the figure indicate number of subjects at risk.
NC = Not Calculable.
The DFS benefit of TAGRISSO compared to placebo was consistent across all predefined subgroups analysed, including ethnicity, age, gender, and EGFR mutation type (Ex19Del or L858R).
Figure 3. Kaplan-Meier curve of overall survival in stage II-IIIA patients:
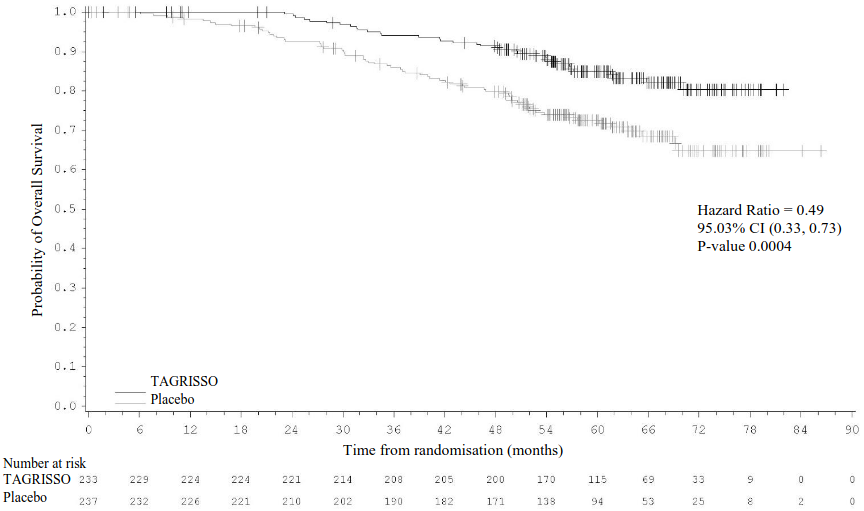
+ Censored patients.
The values at the base of the figure indicate number of subjects at risk.
Figure 4. Kaplan-Meier curve of overall survival in stage IB-IIIA (overall population) patients:
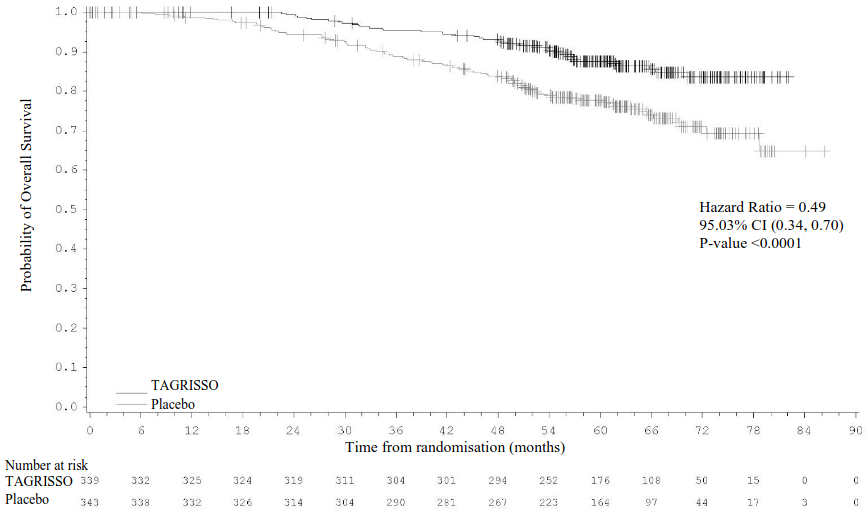
+ Censored patients.
The values at the base of the figure indicate number of subjects at risk.
An exploratory analysis of CNS DFS (time to CNS recurrence or death) for patients on TAGRISSO compared to patients on placebo showed a HR of 0.18 (95% CI: 0.10, 0.33; p<0.0001) for the overall population (stage IB-IIIA).
Patient-reported outcomes
Health-related quality of life (HRQL) in ADAURA was assessed using the Short Form (36) Health Survey version 2 (SF-36v2) questionnaire. SF-36v2 was administered at 12 weeks, 24 weeks and then every 24 weeks relative to randomisation until treatment completion or discontinuation. Overall, HRQL was maintained in both arms up to 30 months, with at least 70% of patients in the stage II-IIIA population not experiencing a clinically meaningful deterioration in the physical component of the SF-36 or death (70% vs 76% for TAGRISSO vs placebo), or in the mental component of the SF-36 or death (70% vs 71% for TAGRISSO vs placebo).
Locally advanced, unresectable EGFR mutation-positive NSCLC – LAURA
The efficacy and safety of TAGRISSO for the treatment of patients with EGFR mutation-positive, locally advanced, unresectable NSCLC, who had not progressed during or following definitive platinum-based chemoradiation therapy, were evaluated in a randomised, double-blind, placebo-controlled study (LAURA). Patients were to receive concurrent chemoradiation therapy (CCRT) or sequential chemoradiation therapy (SCRT) regimens, where at least 2 cycles or 5 weekly doses of platinum-based chemotherapy and a total dose of radiation of 60 Gy ±10% (54 Gy to 66 Gy), were to be completed ≤6 weeks prior to randomisation. Patient tumour tissue samples were required to have an EGFR exon 19 deletion or exon 21 L858R mutation, as identified by central or local testing using a certified/approved test.
Patients were randomised (2:1) to receive either TAGRISSO 80 mg orally once daily (n=143) or placebo (n=73). Randomisation was stratified by prior chemoradiation strategy (CCRT vs SCRT), tumour staging prior to chemoradiation (IIIA vs IIIB/IIIC), and by the China cohort. Patients continued to receive study treatment until intolerance to therapy or confirmed disease progression. Cross-over from placebo to TAGRISSO was permitted upon progressive disease.
The primary efficacy endpoint was progression-free survival (PFS) as assessed by blinded independent central review (BICR). Additional efficacy endpoints included OS and CNS PFS as assessed by neuroradiologist BICR.
The baseline demographic and disease characteristics of the overall study population were: median age 63 years (range 36-84 years), ≥75 years old (13%), female (61%), Asian (82%), White (14%), never smokers (70%). Baseline WHO performance status was 0 (51%) or 1 (49%); 35% of patients had stage IIIA, 49% of patients had stage IIIB and 16% of patients had stage IIIC NSCLC. With regard to EGFR mutation status, 54% were exon 19 deletions and 45% were exon 21 L858R mutations. Prior to randomisation, 89% of patients received CCRT and 11% of patients received SCRT. All patients received platinum-based chemotherapy (55% carboplatin-based chemotherapy and 44% cisplatin-based chemotherapy). The median total dose of radiation was 60 Gy for patients in both arms.
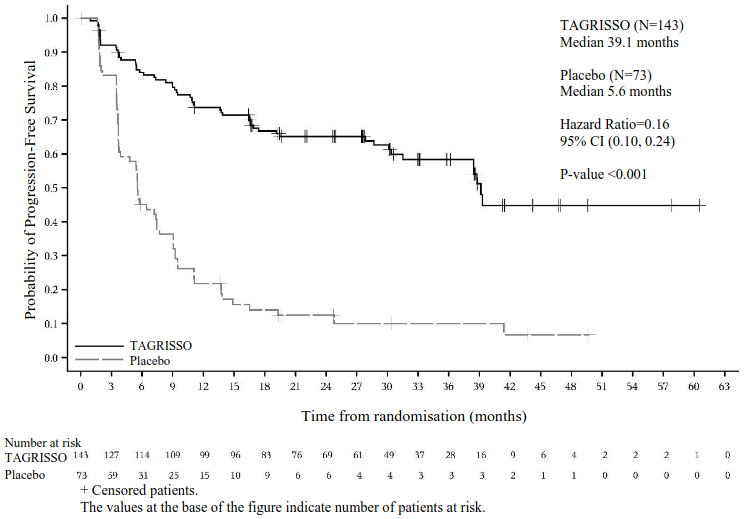
Treatment with TAGRISSO following platinum-based chemoradiation therapy resulted in a
statistically significant improvement in PFS compared to placebo (56% maturity; HR=0.16; 95% CI: 0.10, 0.24; P<0.001, median 39.1 months and 5.6 months, respectively).
Per protocol, all patients underwent baseline magnetic resonance imaging (MRI) brain scans and all but one patient had scheduled on-treatment MRI brain scans. A lower proportion of patients had new CNS lesions by neuroradiologist review in the TAGRISSO arm compared to the placebo arm (17/143 [12%] vs. 26/73 [36%], respectively).
At the time of the interim analysis of OS (DCO: 05 January 2024), statistical significance was not reached. Fifty out of 62 patients (80.6%) in placebo arm were treated with TAGRISSO post-BICR confirmed disease progression.
Efficacy results from LAURA are summarised in Table 4, and the Kaplan-Meier curve for PFS is shown in Figure 5.
Table 4. Efficacy results from LAURA:
| Efficacy Parameter | TAGRISSO (N=143) | Placebo (N=73) |
|---|---|---|
| Progression-Free Survivala | ||
| Number (%) of events | 57 (40) | 63 (86) |
| Median PFS, months (95% CI) | 39.1 (31.5, NC) | 5.6 (3.7, 7.4) |
| HR (95% CI); P-value | 0.16 (0.10, 0.24); P<0.001 | |
| Overall Survival | ||
| Number (%) of deaths | 28 (20) | 15 (21) |
| Median OS, months (95% CI) | 54.0 (46.5, NC) | NC (42.1, NC) |
| HR (95% CI); P-value | 0.81 (0.42, 1.56); P=0.530a | |
HR=Hazard Ratio; CI=Confidence Interval, NC=Not Calculable
PFS results as assessed by BICR.
Median follow-up time for PFS in all patients was 22.0 months in the TAGRISSO arm and 5.6 months in the placebo arm.
a Adjusted for an interim analysis (20% maturity) a p-value <0.000036 was required to achieve statistical significance.
Figure 5. Kaplan-Meier Curves of Progression-Free Survival as assessed by BICR in LAURA:
Previously untreated EGFR mutation-positive locally advanced or metastatic NSCLC
FLAURA – Monotherapy
The efficacy and safety of TAGRISSO for the treatment of patients with EGFR mutation-positive locally advanced, not amenable to curative surgery or radiotherapy, or metastatic NSCLC, who had not received previous systemic treatment for advanced disease, was demonstrated in a randomised, double-blind, active-controlled study (FLAURA). Patient tumour tissue samples were required to have one of the two common EGFR mutations known to be associated with EGFR TKI sensitivity (Ex19del or L858R), as identified by local or central testing.
Patients were randomised 1:1 to receive either TAGRISSO (n=279, 80 mg orally once daily) or EGFR TKI comparator (n=277; gefitinib 250 mg orally once daily or erlotinib 150 mg orally once daily). Randomisation was stratified by EGFR mutation type (Ex19del or L858R) and ethnicity (Asian or non-Asian). Patients received study therapy until intolerance to therapy, or the investigator determined that the patient was no longer experiencing clinical benefit. For patients receiving EGFR TKI comparator, post-progression crossover to open-label TAGRISSO was permitted provided tumour samples tested positive for the T790M mutation. The primary efficacy endpoint was progression-free survival (PFS) as assessed by investigator.
The baseline demographic and disease characteristics of the overall study population were: median age 64 years (range 26-93 years), ≥75 years old (14%), female (63%), White (36%), Asian (62%), never smokers (64%), WHO performance status of 0 or 1 (100%), metastatic bone disease (36%), extra-thoracic visceral metastases (35%), CNS metastases (21%, identified by CNS lesion site at baseline, medical history, and/or prior surgery, and/or prior radiotherapy to CNS metastases).
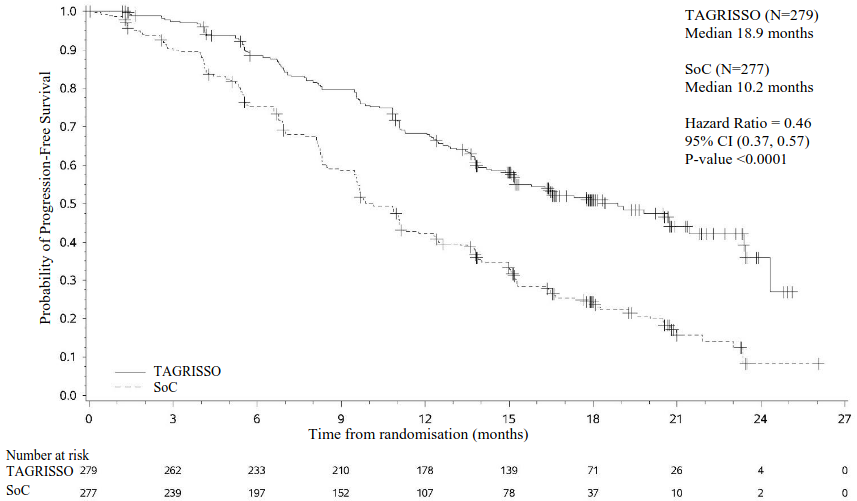
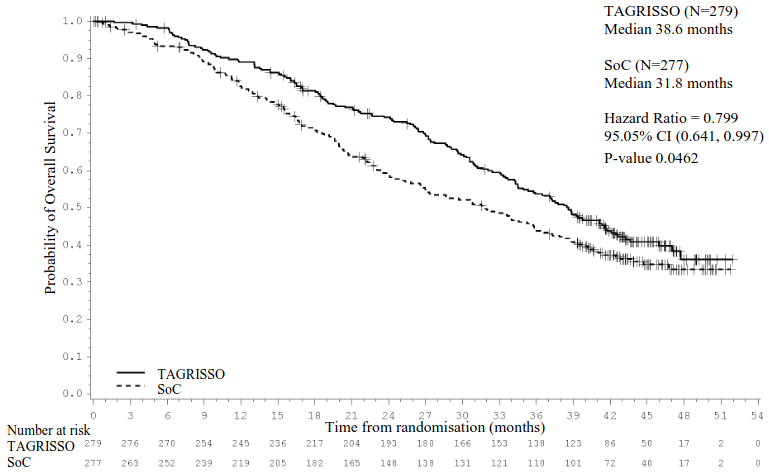
TAGRISSO demonstrated a clinically meaningful and statistically significant improvement in PFS compared to EGFR TKI comparator (median 18.9 months and 10.2 months, respectively, HR=0.46, 95% CI: 0.37, 0.57; P<0.0001). Efficacy results from FLAURA by investigator assessment are summarised in Table 5, and the Kaplan-Meier curve for PFS is shown in Figure 6. The final analysis of OS (58% maturity) demonstrated a statistically significant improvement with an HR of 0.799 (95.05% CI: 0.641, 0.997) and a clinically meaningful longer median survival time in patients randomised to TAGRISSO compared to EGFR TKI comparator (Table 5 and Figure 7). A greater proportion of patients treated with TAGRISSO were alive at 12, 18, 24 and 36 months (89%, 81%, 74% and 54% respectively) compared to patients treated with EGFR TKI comparator (83%, 71%, 59% and 44% respectively). Analysis of post-progression endpoints demonstrated that the PFS benefit was preserved through subsequent lines of therapy.
Table 5. Efficacy results from FLAURA by investigator assessment:
| Efficacy parameter | TAGRISSO (N=279) | EGFR TKI comparator (gefitinib or erlotinib) (N=277) |
|---|---|---|
| Progression-free survival | ||
| Number of events (62% maturity) | 136 (49) | 206 (74) |
| Median PFS, months (95% CI) | 18.9 (15.2, 21.4) | 10.2 (9.6, 11.1) |
| HR (95% CI); P-value | 0.46 (0.37, 0.57); P<0.0001 | |
| Overall survival | ||
| Number of deaths, (58% maturity) | 155 (56) | 166 (60) |
| Median OS, months (95% CI) 3 | 8.6 (34.5, 41.8) | 31.8 (26.6, 36.0) |
| HR (95.05% CI); P-value | 0.799 (0.641, 0.997); P=0.0462a | |
| Objective response rate*b | ||
| Number of responses (n), Response Rate (95% CI) | 223 80% (75, 85) | 210 76% (70, 81) |
| Odds ratio (95% CI); P-value | 1.3 (0.9, 1.9); P=0.2421 | |
| Duration of response (DoR)* | ||
| Median DoR, months (95% CI) | 17.2 (13.8, 22.0) | 8.5 (7.3, 9.8) |
| Second PFS after start of first subsequent therapy (PFS2) | ||
| Number of patients with second progression (%) | 73 (26) | 106 (38) |
| Median PFS2, months (95% CI) | NC (23.7, NC) | 20.0 (18.0, NC) |
| HR (95% CI); P-value | 0.58 (0.44, 0.78); P=0.0004 | |
| Time from randomisation to first subsequent treatment or death (TFST) | ||
| Number of patients who had first subsequent treatment or died (%) | 115 (41) | 175 (63) |
| Median TFST, months (95% CI) | 23.5 (22.0, NC) | 13.8 (12.3, 15.7) |
| HR (95% CI); P-value | 0.51 (0.40, 0.64); P<0.0001 | |
| Time from randomisation to second subsequent treatment or death (TSST) | ||
| Number of patients who had second subsequent treatment or died (%) | 75 (27) | 110 (40) |
| Median TSST, months (95% CI) | NC (NC, NC) | 25.9 (20.0, NC) |
| HR (95% CI); P-value | 0.60 (0.45, 0.80); P=0.0005 | |
HR=Hazard Ratio; CI=Confidence Interval, NC=Not Calculable
PFS, ORR, DoR and PFS2 results based on RECIST investigator assessment.
* Based on unconfirmed response.
Median follow-up time was 15.0 months for patients receiving TAGRISSO and 9.7 months for patients receiving EGFR TKI comparator.
Median survival follow-up time was 35.8 months for patients receiving TAGRISSO and 27.0 months for patients receiving EGFR TKI comparator.
PFS, ORR, DoR, PFS2, TFST and TSST results are from DCO 12 June 2017. OS results are from DCO 25 June 2019.
A HR <1 favours TAGRISSO, an Odds ratio of >1 favours TAGRISSO.
a Adjusted for an interim analysis (25% maturity) a p-value <0.0495 was required to achieve statistical significance.
b ORR results by Blinded Independent Central Review (BICR) were consistent with those reported via investigator assessment; ORR by BICR assessment was 78% (95% CI: 73, 83) on TAGRISSO and 70% (95% CI: 65, 76) on EGFR TKI comparator.
Figure 6. Kaplan-Meier curves of progression-free survival as assessed by investigator in FLAURA:
+ Censored patients.
The values at the base of the figure indicate number of subjects at risk.
Figure 7. Kaplan-Meier curves of overall survival in FLAURA:
+ Censored patients.
The values at the base of the figure indicate number of subjects at risk.
The PFS benefit of TAGRISSO compared to EGFR TKI comparator was consistent across all predefined subgroups analysed, including ethnicity, age, gender, smoking history, CNS metastases status at study entry and EGFR mutation type (Exon 19 deletion or L858R).
CNS metastases efficacy data in FLAURA study
Patients with CNS metastases not requiring steroids and with stable neurologic status for at least two weeks after completion of the definitive therapy and steroids were eligible to be randomised in the FLAURA study. Of 556 patients, 200 patients had available baseline brain scans. A Blinded Independent Central Review (BICR) assessment of these scans resulted in a subgroup of 128/556 (23%) patients with CNS metastases and these data are summarised in Table 6. CNS efficacy by RECIST v1.1 in FLAURA demonstrated a statistically significant improvement in CNS PFS (HR=0.48, 95% CI 0.26, 0.86; P=0.014).
Table 6. CNS efficacy by BICR in patients with CNS metastases on a baseline brain scan in FLAURA:
| Efficacy parameter | TAGRISSO N=61 | EGFR TKI comparator (gefitinib or erlotinib) N=67 |
|---|---|---|
| CNS progression-free survivala | ||
| Number of events (%) 1 | 8 (30) | 30 (45) |
| Median CNS PFS, months (95% CI) | NC (16.5, NC) | 13.9 (8.3, NC) |
| HR (95% CI); P-value | 0.48 (0.26, 0.86); P=0.014 | |
| CNS progression free and alive at 6 months (%) (95% CI) | 87 (74, 94) | 71 (57, 81) |
| CNS progression free and alive at 12 months (%) (95% CI) | 77 (62, 86) | 56 (42, 68) |
HR=Hazard Ratio; CI=Confidence Interval, NC=Not Calculable
A HR <1 favours TAGRISSO, an Odds ratio of >1 favours TAGRISSO
a CNS PFS determined by RECIST v1.1 by CNS BICR (CNS measurable and non-measurable lesions at baseline by BICR) n=61 for TAGRISSO and n=67 for EGFR TKI comparator; responses are unconfirmed.
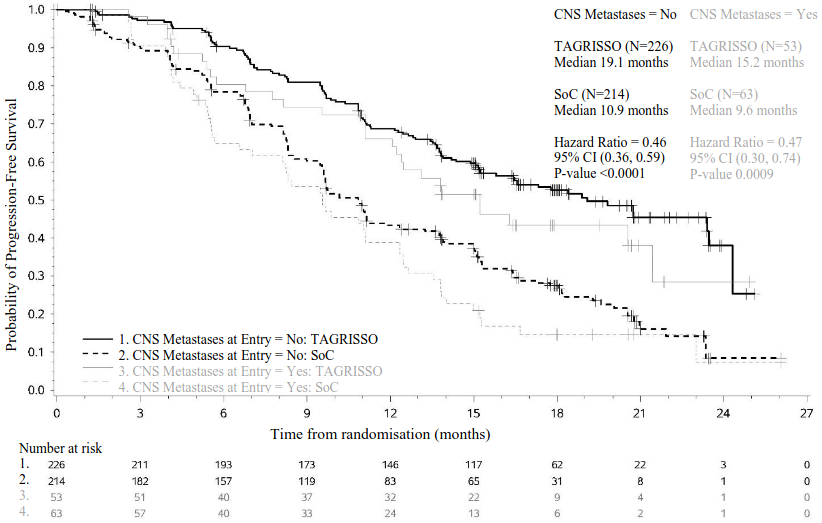
A pre-specified PFS subgroup based on CNS metastases status (identified by CNS lesion site at baseline, medical history, and/or prior surgery, and/or prior radiotherapy to CNS metastases) at study entry was performed in FLAURA and is shown in Figure 8. Irrespective of CNS lesion status at study entry, patients in the TAGRISSO arm demonstrated an efficacy benefit over those in the EGFR TKI comparator arm and there were fewer patients with new CNS lesions in the TAGRISSO arm compared to the EGFR TKI comparator arm (TAGRISSO, 11/279 [3.9%] compared to EGFR TKI comparator, 34/277 [12.3%]). In the subset of patients without CNS lesions at baseline, there were a lower number of new CNS lesions in the TAGRISSO arm compared to the EGFR TKI comparator arm (7/226 [3.1%] vs. 15/214 [7.0%], respectively).
Figure 8. Overall PFS by investigator assessment by CNS metastases status at study entry, Kaplan- Meier plot (full analysis set) in FLAURA:
+ Censored patients.
The values at the base of the figure indicate number of subjects at risk.
Patient-reported outcomes
Patient-reported symptoms and HRQL were electronically collected using the EORTC QLQ-C30 and its lung cancer module (EORTC QLQ-LC13). The LC13 was initially administered once a week for the first 6 weeks, then every 3 weeks before and after progression. The C30 was assessed every 6 weeks before and after progression. At baseline, no differences in patient reported symptoms, function or HRQL were observed between TAGRISSO and EGFR TKI comparator (gefitinib or erlotinib) arms. Compliance over the first 9 months was generally high (≥70%) and similar in both arms.
Key lung cancer symptoms analysis:
Data collected from baseline up to month 9 showed similar improvements in TAGRISSO and EGFR TKI comparator groups for the five pre-specified primary PRO symptoms (cough, dyspnoea, chest pain, fatigue, and appetite loss) with improvement in cough reaching the established clinically relevant cut-off. Up to month 9 there were no clinically meaningful differences in patient-reported symptoms between TAGRISSO and EGFR TKI comparator groups (as assessed by a difference of ≥10 points).
HRQL and physical functioning improvement analysis:
Both groups reported similar improvements in most functioning domains and global health status/HRQL, indicating that patients' overall health status improved. Up to month 9, there were no clinically meaningful differences between the TAGRISSO and EGFR TKI comparator groups in functioning or HRQL.
FLAURA2 – Combination Therapy
The efficacy and safety of TAGRISSO in combination with pemetrexed and platinum-based chemotherapy for the treatment of patients with EGFR mutation-positive locally advanced or metastatic NSCLC, who had not received previous systemic treatment for advanced disease, was demonstrated in a randomised, open-label, active-controlled study (FLAURA2). Patient tumour tissue samples were required to have one of the two common EGFR mutations known to be associated with EGFR TKI sensitivity (Ex19del or L858R), as identified by local or central testing.
Patients were randomised (1:1) to one of the following treatment arms:
- TAGRISSO (80 mg) orally once daily with pemetrexed (500 mg/m²) and the investigator's choice of cisplatin (75 mg/m²) or carboplatin (AUC5) administered intravenously on Day 1 of every 21-day cycle for 4 cycles, followed by TAGRISSO (80 mg) orally once daily and pemetrexed (500 mg/m²) administered intravenously every 3 weeks (n=279)
- TAGRISSO (80 mg) orally once daily (n=278)
Randomisation was stratified by race (Chinese/Asian, non-Chinese/Asian or non-Asian), WHO performance status (0 or 1), and method for tissue testing (central or local). Patients received study therapy until intolerance to therapy, or the investigator determined that the patient was no longer experiencing clinical benefit.
The primary efficacy endpoint was PFS as assessed by investigator per RECIST 1.1 and the key secondary efficacy endpoint was OS.
The baseline demographic and disease characteristics of the overall study population were: median age 61 years (range 26-85 years), ≥75 years old (8%), female (61%), Asian (64%), White (28%), never smokers (66%). Baseline WHO performance status was 0 (37%) or 1 (63%); 98.7% had predominantly adenocarcinoma histology. Of the patients who had metastatic disease, 49% had metastatic bone disease, 53% had extra-thoracic metastases and 20% had liver metastases. Forty-one percent (41%) of patients had CNS metastases (identified by investigator based on CNS lesion site at baseline, medical history, and/or prior surgery, and/or prior radiotherapy to CNS metastases). With regard to tumour EGFR mutation type at randomisation, 60.5% were exon 19 deletions and 38.2% were exon 21 L858R; 0.7% of patients had tumours with both exon 19 deletions and exon 21 L858R.
TAGRISSO in combination with pemetrexed and platinum-based chemotherapy demonstrated a statistically significant improvement in PFS compared to TAGRISSO monotherapy. The PFS benefit was consistent across all subgroups analysed. At the time of the second interim analysis of OS (DCO 08 January 2024), statistical significance was not reached.
Efficacy results from FLAURA2 by investigator assessment are summarised in Table 7, the Kaplan-Meier curve for PFS is shown in Figure 9 and the Kaplan-Meier curve for OS is shown in Figure 10.
Table 7. Efficacy results from FLAURA2 by investigator assessment:
| Efficacy parameter | TAGRISSO with pemetrexed and platinum- based chemotherapy (N=279) | TAGRISSO (N=278) |
|---|---|---|
| Progression-Free Survival | ||
| Number (%) of events | 120 (43) | 166 (60) |
| Median PFS, months (95% CI)a | 25.5 (24.7, NC) | 16.7 (14.1, 21.3) |
| HR (95% CI); P-value | 0.62 (0.49, 0.79); P<0.0001 | |
| Overall Survival | ||
| Number (%) of deaths | 100 (36) | 126 (45) |
| Median OS, months (95% CI) | NC (38.0, NC) | 36.7 (33.2, NC) |
| HR (95% CI); P-value | 0.75 (0.57, 0.97); P=0.0280b | |
HR=Hazard Ratio; CI=Confidence Interval, NC=Not Calculable
PFSbased on RECIST investigator assessment.
Median follow-up time for PFS in all patients was 19.5 months in the TAGRISSO with pemetrexed and platinum-based chemotherapy arm and 16.5 months in the TAGRISSO monotherapy arm.
PFS results are from DCO 03 April 2023 (51% maturity). OS results are from DCO 08 January 2024 (41% maturity).
a PFS results by BICR were consistent with those reported via investigator assessment.
b Based on the second interim analysis (41% maturity) a p-value <0.000001 was required to achieve statistical significance.
Figure 9. Kaplan-Meier curves of progression-free survival as assessed by investigator in FLAURA2:
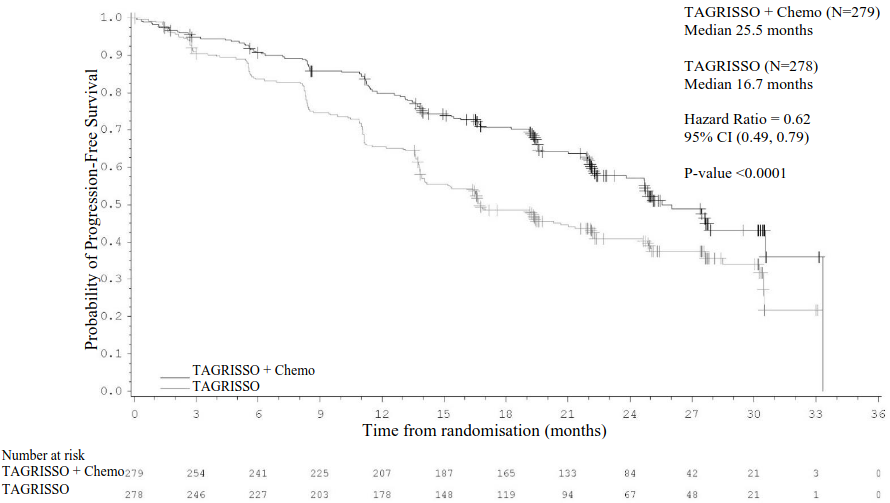
+ Censored patients.
Chemo = Pemetrexed and platinum-based chemotherapy.
The values at the base of the figure indicate number of patients at risk.
Figure 10. Kaplan-Meier Curves of Overall Survival in FLAURA2:
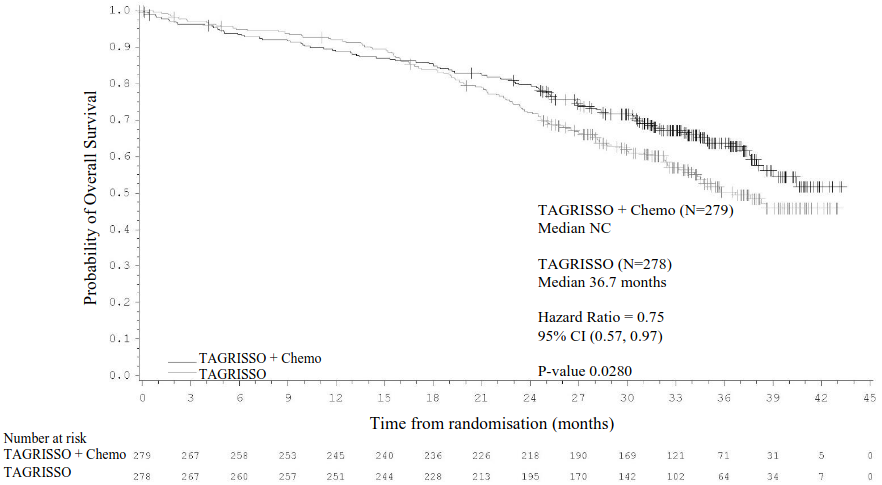
+ Censored patients.
Chemo = Pemetrexed and platinum-based chemotherapy.
The values at the base of the figure indicate number of patients at risk.
CNS metastases efficacy data in FLAURA2 study
Patients with asymptomatic CNS metastases not requiring steroids and with stable neurologic status for at least two weeks after completion of the definitive therapy and steroids were eligible to be randomised in the FLAURA2 study. All patients had available baseline brain scans. A BICR assessment, using modified RECIST, of these scans resulted in a subgroup of 222/557 (40%) patients with CNS measurable and/or non-measurable lesions (cFAS) and a further subgroup of 78/557 (14%) patients with CNS measurable lesions (cEFR). Based on an exploratory analysis, CNS response rate was >65% in both treatment arms with a higher complete response rate in the TAGRISSO with pemetrexed and platinum-based chemotherapy arm (59.3% of patients) compared to the TAGRISSO monotherapy arm (43.3% of patients). Median DoR was not reached in the TAGRISSO with pemetrexed and platinum-based chemotherapy arm and was 26.2 months in the TAGRISSO monotherapy arm. In the cEFR subgroup, 47.5% of patients in the TAGRISSO with pemetrexed and platinum-based chemotherapy arm had a CNS complete response compared to 15.8% of patients in the TAGRISSO monotherapy arm.
Pre-treated T790M-positive NSCLC patients-AURA3
The efficacy and safety of TAGRISSO for the treatment of patients with locally advanced or metastatic T790M NSCLC whose disease has progressed on or after EGFR TKI therapy, was demonstrated in a randomised, open-label, active-controlled Phase 3 study (AURA3). All patients were required to have EGFR T790M mutation-positive NSCLC identified by the cobas EGFR mutation test performed in a central laboratory prior to randomisation. The T790M mutation status was also assessed using ctDNA extracted from a plasma sample taken during screening. The primary efficacy outcome was PFS as assessed by investigator. Additional efficacy outcome measures included ORR, DoR and OS as assessed by investigator.
Patients were randomised in a 2:1 (TAGRISSO: platinum-based doublet chemotherapy) ratio to receive TAGRISSO (n=279) or platinum-based doublet chemotherapy (n=140). Randomisation was stratified by ethnicity (Asian and non-Asian). Patients in the TAGRISSO arm received TAGRISSO 80 mg orally once daily until intolerance to therapy, or the investigator determined that the patient was no longer experiencing clinical benefit. Chemotherapy consisted of pemetrexed 500 mg/m² with carboplatin AUC5 or pemetrexed 500 mg/m² with cisplatin 75 mg/m² on Day 1 of every 21-day cycle for up to 6 cycles. Patients whose disease has not progressed after four cycles of platinum-based chemotherapy may receive pemetrexed maintenance therapy (pemetrexed 500 mg/m² on Day 1 of every 21-day cycle). Subjects on the chemotherapy arm who had objective radiological progression (by the investigator and confirmed by independent central imaging review) were given the opportunity to begin treatment with TAGRISSO.
The baseline demographic and disease characteristics of the overall study population were: median age 62, ≥75 years old (15%), female (64%), white (32%), Asian (65%), never smoker (68%), WHO performance status 0 or 1 (100%). Fifty-four percent (54%) of patients had extra-thoracic visceral metastases, including 34% with CNS metastases (identified by CNS lesion site at baseline, medical history, and/or prior surgery, and/or prior radiotherapy to CNS metastases) and 23% with liver metastases. Forty-two percent (42%) of patients had metastatic bone disease.
AURA3 demonstrated a statistically significant improvement in PFS in the patients treated with TAGRISSO compared to chemotherapy. Efficacy results from AURA3 by investigator assessment are summarised in Table 8, and the Kaplan-Meier curve for PFS is shown in Figure 11. No statistically significant difference was observed between the treatment arms at the final OS analysis.
Table 8. Efficacy results from AURA3 by investigator assessment:
| Efficacy parameter | TAGRISSO (N=279) | Chemotherapy (Pemetrexed/Cisplatin or Pemetrexed/Carboplatin) (N=140) |
|---|---|---|
| Progression-free survival | ||
| Number of events (% maturity) | 140 (50) | 110 (79) |
| Median PFS, months (95% CI) | 10.1 (8.3, 12.3) | 4.4 (4.2, 5.6) |
| HR (95% CI); P-value | 0.30 (0.23,0.41); P<0.001 | |
| Overall survival (OS)a | ||
| Number of deaths (% maturity) | 188 (67.4) | 93 (66.4) |
| Median OS, months (95% CI) | 26.8 (23.5, 31.5) | 22.5 (20.2, 28.8) |
| HR (95.56% CI); P-value | 0.87 (0.67, 1.13); P=0.277 | |
| Objective response rateb | ||
| Number of responses, response rate (95% CI) | 197 71% (65, 76) | 44 31% (24, 40) |
| Odds ratio (95% CI); P-value | 5.4 (3.5, 8.5); P<0.001 | |
| Duration of response (DoR)b | ||
| Median DoR, months (95% CI) | 9.7 (8.3, 11.6) | 4.1 (3.0, 5.6) |
HR=Hazard Ratio; CI=confidence interval; NC=non-calculable; OS=Overall Survival
All efficacy results based on RECIST investigator assessment.
a The final analysis of OS was performed at 67% maturity. The CI for the HR has been adjusted for previous interim analyses. The OS analysis was not adjusted for the potentially confounding effects of crossover (99 [71%] patients on the chemotherapy arm received subsequent osimertinib treatment).
b ORR and DoR results by investigator assessment were consistent with those reported via BICR; ORR by BICR assessment was 64.9% [95% CI: 59.0, 70.5] on osimertinib and 34.3% [95% CI: 25.6, 42.8] on chemotherapy; DoR by BICR assessment was 11.2 months (95% CI: 8.3, NC) on osimertinib and 3.1 months (95% CI: 2.9, 4.3) on chemotherapy.
Figure 11. Kaplan-Meier curves of progression-free survival as assessed by investigator in AURA3:
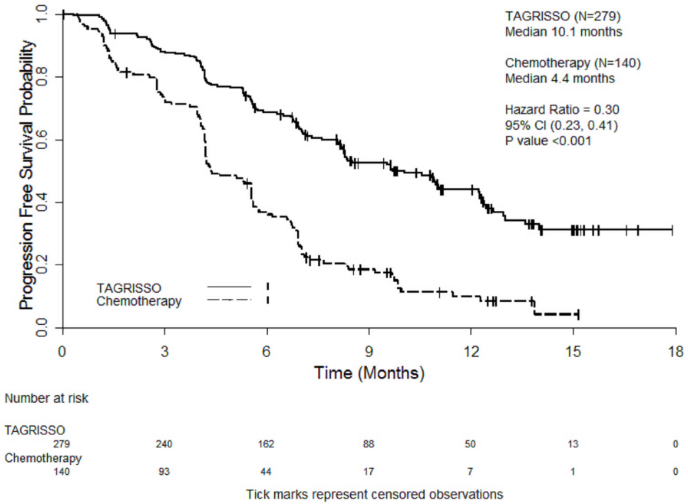
A sensitivity analysis of PFS was conducted by a BICR and showed a median PFS of 11.0 months with TAGRISSO compared with 4.2 months with chemotherapy. This analysis demonstrated a consistent treatment effect (HR 0.28; 95% CI: 0.20, 0.38) with that observed by investigator assessment.
Clinically meaningful improvements in PFS with HRs less than 0.50 in favour of patients receiving TAGRISSO compared to those receiving chemotherapy were consistently observed in all predefined subgroups analysed, including ethnicity, age, gender, smoking history and EGFR mutation (Exon 19 deletion and L858R).
CNS metastases efficacy data in AURA3 study
Patients with asymptomatic, stable brain metastases not requiring steroids for at least 4 weeks prior to the start of study treatment were eligible to be randomised in the study. A BICR assessment of CNS efficacy by RECIST v1.1 in the subgroup of 116/419 (28%) patients identified to have CNS metastases on a baseline brain scan are summarised in Table 9.
Table 9. CNS efficacy by BICR in patients with CNS metastases on a baseline brain scan in AURA3:
| Efficacy parameter | TAGRISSO | Chemotherapy (Pemetrexed/Cisplatin or Pemetrexed/Carboplatin) |
|---|---|---|
| CNS objective response ratea | ||
| CNS response rate % (n/N) (95% CI) | 70% (21/30) (51, 85) | 31% (5/16) (11%, 59%) |
| Odds ratio (95% CI); P-value | 5.1 (1.4, 21); P=0.015 | |
| CNS duration of responseb | ||
| Median CNS DoR, months (95% CI) | 8.9 (4.3, NC) | 5.7 (NC, NC) |
| CNS disease control rate | ||
| CNS disease control rate | 87% (65/75) (77, 93) | 68% (28/41) (52, 82) |
| Odds ratio (95% CI); P-value | 3 (1.2, 7.9); P=0.021 | |
| CNS progression-free survivalc | N=75 | N=41 |
| Number of events (% maturity) | 19 (25) | 16 (39) |
| Median CNS PFS, months (95% CI) | 11.7 (10, NC) | 5.6 (4.2, 9.7) |
| HR (95% CI); P-value | 0.32 (0.15, 0.69); P=0.004 | |
a CNS Objective Response Rate and Duration of Response determined by RECIST v1.1 by CNS BICR in the evaluable for response population (CNS measurable lesions at baseline by BICR) n=30 for TAGRISSO and n=16 for Chemotherapy.
b Based on patients with response only; DoR defined as the time from the date of first documented response (complete response or partial response) until progression or death event; DCR defined as the proportion of patients with response (complete response or partial response), or stable disease ≥6 weeks.
c CNS Progression Free Survival determined by RECIST v1.1 by CNS BICR in the full analysis set population (CNS measurable and non-measurable lesions at baseline by BICR) n=75 for TAGRISSO and n=41 for Chemotherapy.
A HR <1 favours TAGRISSO.
A pre-specified PFS subgroup analysis based on CNS metastases status at study entry was performed in AURA3 and is shown in Figure 12.
Figure 12. Overall PFS by investigator assessment by CNS metastases status at study entry, Kaplan-Meier plot (full analysis set) in AURA3:
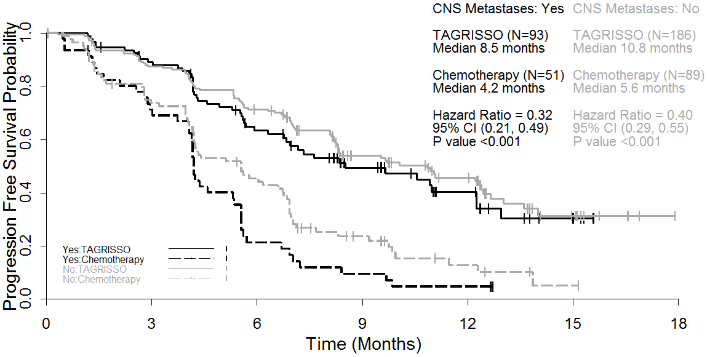
AURA3 demonstrated a statistically significant improvement in PFS for patients receiving TAGRISSO compared to those receiving chemotherapy irrespective of CNS metastases status at study entry.
Patient-reported outcomes
Patient-reported symptoms and HRQL were electronically collected using the EORTC QLQ-C30 and its lung cancer module (EORTC QLQ-LC13). The LC13 was initially administered once a week for the first 6 weeks, then every 3 weeks before and after progression. The C30 was assessed every 6 weeks before and after progression.
Key lung cancer symptoms analysis:
TAGRISSO improved patient-reported lung cancer symptoms compared to chemotherapy by demonstrating a statistically significant difference in mean change from baseline versus chemotherapy during the overall time period from randomisation until 6 months for 5 pre-specified primary PRO symptoms (appetite loss, cough, chest pain, dyspnoea, and fatigue) as shown in Table 10.
Table 10. Mixed Model Repeated Measures – Key lung cancer symptoms - mean change from baseline in TAGRISSO patients compared with chemotherapy:
| Appetite Loss | Cough | Chest Pain | Dyspnoea | Fatigue | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Arms | TAGRISSO (279) | Chemo- therapy (140) | TAGRISSO (279) | Chemo- therapy (140) | TAGRISSO (279) | Chemo- therapy (140) | TAGRISSO (279) | Chemo- therapy (140) | TAGRISSO (279) | Chemo- therapy (140) |
| N | 239 | 97 | 228 | 113 | 228 | 113 | 228 | 113 | 239 | 97 |
| Adj Mean | -5.51 | 2.73 | -12.22 | -6.69 | -5.15 | 0.22 | -5.61 | 1.48 | -5.68 | 4.71 |
| Estimated Difference (95%CI) | -8.24 (-12.88, 3.60) | -5.53 (-8.89, -2.17) | -5.36 (-8.20, -2.53) | -7.09 (-9.86, -4.33) | -10.39 (-14.55, -6.23) | |||||
| p-value | p<0.001 | p=0.001 | p<0.001 | p<0.001 | p<0.001 | |||||
Adjusted mean and estimated differences obtained from a Mixed Model Repeated Measures (MMRM) analysis. The model included patient, treatment, visit, treatment-by-visit interaction, baseline symptom score, and baseline symptom score-by-visit interaction and used an unstructured covariance matrix.
HRQL and physical functioning improvement analysis:
Patients on TAGRISSO had significantly greater chances of achieving a clinically meaningful improvement of greater than or equal to 10 points on the global health status and physical functioning of the EORTC-C30 questionnaire compared with chemotherapy during the study period Odds Ratio (OR) global health status: 2.11, (95% CI 1.24, 3.67, p=0.007); OR physical functioning 2.79 (95% CI 1.50, 5.46, p=0.002).
Pre-treated T790M-positive NSCLC patients - AURAex and AURA2
Two single-arm, open-label clinical studies, AURAex (Phase 2 Extension cohort, (n=201)) and AURA2 (n=210) were conducted in patients with EGFR T790M mutation-positive lung cancer who have progressed on one or more prior systemic therapies, including an EGFR TKI. All patients were required to have EGFR T790M mutation-positive NSCLC identified by the cobas EGFR mutation test performed in a central laboratory prior to treatment. The T790M mutation status was also assessed retrospectively using ctDNA extracted from a plasma sample taken during screening. All patients received TAGRISSO at a dose of 80 mg once daily. The primary efficacy outcome measure of these two studies was ORR according to RECIST v1.1 as evaluated by a BICR. Secondary efficacy outcome measures included DoR and PFS.
Baseline characteristics of the overall study population (AURAex and AURA2) were as follows: median age 63 years, 13% of patients were ≥75 years old, female (68%), White (36%), Asian (60%). All patients received at least one prior line of therapy. Thirty-one percent (31%) (N=129) had received 1 prior line of therapy (EGFR-TKI treatment only), 69% (N=282) had received 2 or more prior lines. Seventy-two percent (72%) of patients were never smokers, 100% of patients had a WHO performance status of 0 or 1. Fifty-nine percent (59%) of patients had extra-thoracic visceral metastasis including 39% with CNS metastases (identified by CNS lesion site at baseline, medical history, and/or prior surgery and/or prior radiotherapy to CNS metastases) and 29% with liver metastases. Forty-seven percent (47%) of patients had metastatic bone disease. The median duration of follow up for PFS was 12.6 months.
In the 411 pre-treated EGFR T790M mutation-positive patients, the total ORR by BICR was 66% (95% CI: 61, 71). In patients with a confirmed response by BICR, the median DoR was 12.5 months (95% CI: 11.1, NE). The ORR by BICR in AURAex was 62% (95% CI: 55, 68) and 70% (95% CI: 63, 77) in AURA2. The median PFS was 11.0 months 95% CI (9.6, 12.4).
Objective response rates by BICR above 50% were observed in all predefined subgroups analysed, including line of therapy, ethnicity, age and region.
In the evaluable for response population, 85% (223/262) had documentation of response at the time of the first scan (6 weeks); 94% (247/262) had documentation of response at the time of the second scan (12 weeks).
CNS metastases efficacy data in Phase 2 studies (AURAex and AURA2)
A BICR assessment of CNS efficacy by RECIST v 1.1 was performed in a subgroup of 50 (out of 411) patients identified to have measurable CNS metastases on a baseline brain scan. A CNS ORR of 54% (27/50 patients; 95% CI: 39.3, 68.2) was observed with 12% of these responses being complete responses.
Clinical studies have not been conducted in patients with de novo EGFR T790M mutation-positive NSCLC.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with TAGRISSO in all subsets of the paediatric population in NSCLC (see section 4.2 for information on paediatric use).
Pharmacokinetic properties
Osimertinib pharmacokinetic parameters have been characterised in healthy subjects and NSCLC patients. Based on population pharmacokinetic analysis, osimertinib apparent plasma clearance is 14.3 L/h, apparent volume of distribution is 918 L and terminal half-life of approximately 44 hours. The pharmacokinetics in patients treated with osimertinib in combination with pemetrexed and platinum-based chemotherapy are similar to those in patients treated with osimertinib monotherapy. The AUC and Cmax increased dose proportionally over 20 to 240 mg dose range. Administration of osimertinib once daily results in approximately 3-fold accumulation with steady-state exposures achieved by 15 days of dosing. At steady-state, circulating plasma concentrations are typically maintained within a 1.6-fold range over the 24-hour dosing interval.
Absorption
Following oral administration of TAGRISSO, peak plasma concentrations of osimertinib were achieved with a median (min-max) tmax of 6 (3-24) hours, with several peaks observed over the first 24 hours in some patients. The absolute bioavailability of TAGRISSO is 70% (90% CI 67, 73). Based on a clinical pharmacokinetic study in patients at 80 mg, food does not alter osimertinib bioavailability to a clinically meaningful extent (AUC increase by 6% (90% CI -5, 19) and Cmax decrease by 7% (90% CI -19, 6)). In healthy volunteers administered an 80 mg tablet where gastric pH was elevated by dosing of omeprazole for 5 days, osimertinib exposure was not affected (AUC and Cmax increase by 7% and 2%, respectively) with the 90% CI for exposure ratio contained within the 80-125% limit.
Distribution
Population estimated mean volume of distribution at steady-state (Vss/F) of osimertinib is 918 L indicating extensive distribution into tissue. In vitro plasma protein binding of osimertinib is 94.7% (5.3% free). Osimertinib has also been demonstrated to bind covalently to rat and human plasma proteins, human serum albumin and rat and human hepatocytes.
Biotransformation
In vitro studies indicate that osimertinib is metabolised predominantly by CYP3A4, and CYP3A5. However, with current available data, alternative metabolic pathways cannot be fully ruled out. Based on in vitro studies, 2 pharmacologically active metabolites (AZ7550 and AZ5104) have subsequently been identified in the plasma of preclinical species and in humans after oral dosing with osimertinib; AZ7550 showed a similar pharmacological profile to TAGRISSO while AZ5104 showed greater potency across both mutant and wild-type EGFR. Both metabolites appeared slowly in plasma after administration of TAGRISSO to patients, with a median (min-max) tmax of 24 (4-72) and 24 (6-72) hours, respectively. In human plasma, parent osimertinib accounted for 0.8%, with the 2 metabolites contributing 0.08% and 0.07% of the total radioactivity with the majority of the radioactivity being covalently bound to plasma proteins. The geometric mean exposure of both AZ5104 and AZ7550, based on AUC, was approximately 10% each of the exposure of osimertinib at steady-state.
The main metabolic pathway of osimertinib was oxidation and dealkylation. At least 12 components were observed in the pooled urine and faecal samples in humans with 5 components accounting for >1% of the dose of which unchanged osimertinib, AZ5104 and AZ7550, accounted for approximately 1.9, 6.6 and 2.7% of the dose while a cysteinyl adduct (M21) and an unknown metabolite (M25) accounted for 1.5% and 1.9% of the dose, respectively.
Based on in vitro studies, osimertinib is a competitive inhibitor of CYP 3A4/5 but not CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6 and 2E1 at clinically relevant concentrations. Based on in vitro studies, osimertinib is not an inhibitor of UGT1A1 and UGT2B7 at clinically relevant concentrations hepatically. Intestinal inhibition of UGT1A1 is possible but the clinical impact is unknown.
Elimination
Following a single oral dose of 20 mg, 67.8% of the dose was recovered in faeces (1.2% as parent) while 14.2% of the administered dose (0.8% as parent) was found in urine by 84 days of sample collection. Unchanged osimertinib accounted for approximately 2% of the elimination with 0.8% in urine and 1.2% in faeces.
Interactions with transport proteins
In vitro studies have shown that osimertinib is not a substrate of OATP1B1 and OATP1B3. In vitro, osimertinib does not inhibit OAT1, OAT3, OATP1B1, OATP1B3, MATE1, OCT2 and MATE2K at clinically relevant concentrations.
Based on in vitro studies osimertinib is a substrate of P-gp and BCRP but at clinical doses, clinically relevant interactions are unlikely. Based on in vitro data, osimertinib is an inhibitor of BCRP and P-gp (see section 4.5).
Special populations
In a population based pharmacokinetic analyses (n=1367), no clinically significant relationships were identified between predicted steady-state exposure (AUCss) and patient's age (range: 25 to 91 years), gender (65% female), ethnicity (including White, Asian, Japanese, Chinese and non-Asian-non-White patients), line of therapy and smoking status (n=34 current smokers, n=419 former smokers). Population PK analysis indicated that body weight was a significant covariate with a less than 20% change in osimertinib AUCss expected across a body weight range of 88 kg to 43 kg respectively (95% to 5% quantiles) when compared to the AUCss for the median body weight of 61 kg. Taking the extremes of body weight into consideration, from <43 kg to >88 kg, AZ5104 metabolite ratios ranged from 11.8% to 9.6% while for AZ7550 it ranged from 12.8% to 8.1%, respectively. Based on population PK analysis, serum albumin was identified as a significant covariate with a <30% change in osimertinib AUCss expected across the albumin range of 29 to 46 g/L respectively (95% to 5% quantiles) when compared to the AUCss for the median baseline albumin of 39 g/L. These exposure changes due to body weight or baseline albumin differences are not considered clinically relevant.
Hepatic impairment
Osimertinib is eliminated mainly via the liver. In a clinical study, patients with different types of advanced solid tumours and with mild hepatic impairment (Child Pugh A, mean score =5.3, n=7) or moderate hepatic impairment (Child Pugh B, mean score =8.2, n=5) had no increase in exposure compared to patients with normal hepatic function (n=10) after a single 80 mg dose of TAGRISSO. The geometric mean ratio (90% CI) of osimertinib AUC and Cmax was 63.3% (47.3, 84.5) and 51.4% (36.6, 72.3) in patients with mild hepatic impairment and 68.4% (49.6, 94.2) and 60.7% (41.6, 88.6) in patients with moderate hepatic impairment; for the metabolite AZ5104 the AUC and Cmax were 66.5% (43.4, 101.9) and 66.3% (45.3, 96.9) in patients with mild hepatic impairment and 50.9% (31.7, 81.6) and 44.0% (28.9, 67.1) in patients with moderate hepatic impairment, compared to the exposure in patients with normal hepatic function. Based on population PK analysis, there was no relationship between markers of hepatic function (ALT, AST, bilirubin) and osimertinib exposure. The hepatic impairment marker serum albumin showed an effect on the PK of osimertinib. Clinical studies that were conducted excluded patients with AST or ALT >2.5x upper limit of normal (ULN), or if due to underlying malignancy, >5.0x ULN or with total bilirubin >1.5x ULN. Based on a pharmacokinetic analysis of 134 patients with mild hepatic impairment, 8 patients with moderate hepatic impairment and 1216 patients with normal hepatic function osimertinib exposures were similar. There are no data available on patients with severe hepatic impairment (see section 4.2).
Renal impairment
In a clinical study, patients with severe renal impairment (CLcr 15 to less than 30 mL/min; n=7) compared to patients with normal renal function (CLcr greater than or equal to 90 mL/min; n=8) after a single 80 mg oral dose of TAGRISSO showed a 1.85-fold increase in AUC (90% CI; 0.94, 3.64) and a 1.19-fold increase in Cmax (90% CI: 0.69, 2.07). Furthermore, based on a population pharmacokinetic analysis of 593 patients with mild renal impairment (CLcr 60 to less than 90 mL/min), 254 patients with moderate renal impairment (CLcr 30 to less than 60 mL/min), 5 patients with severe renal impairment (CLcr 15 to less than 30 mL/min) and 502 patients with normal renal function (greater than or equal to 90 mL/min), osimertinib exposures were similar. Patients with CLcr less than or equal to 10 mL/min were not included in the clinical studies.
Patients with brain metastases
PET images following administration of microdoses of [11C]osimertinib in EGFR mutation-positive NSCLC patients with brain metastases (n=4) and healthy volunteers (n=7) demonstrated that the brain to plasma ratio (Kp) was similar and that [11C]osimertinib crossed the blood brain barrier rapidly and was homogeneously distributed across all regions of the brain in both patients and healthy volunteers.
Preclinical safety data
The main findings observed in repeat dose toxicity studies in rats and dogs comprised atrophic, inflammatory and/or degenerative changes affecting the epithelia of the cornea (accompanied by corneal translucencies and opacities in dogs at ophthalmology examination), GI tract (including tongue), skin, and male and female reproductive tracts with secondary changes in spleen. These findings occurred at plasma concentrations that were below those seen in patients at the 80 mg therapeutic dose. The findings present following 1 month of dosing were largely reversible within 1 month of cessation of dosing with the exception of partial recovery for some of the corneal changes.
Lens fibre degeneration was found in the 104-week carcinogenicity rat study at exposures 0.2 times the human AUC, at the recommended clinical dose of 80 mg once a day. Lens opacities were first noted from week 52 of this study and showed a gradual increase in incidence and severity with increased duration of dosing. The clinical relevance of this finding cannot be ruled out.
Osimertinib penetrated the intact blood-brain barrier of the cynomolgus monkey (intravenous dosing), rat and mouse (oral administration).
Non-clinical data indicate that osimertinib and its metabolite (AZ5104) inhibit the h-ERG channel, and QTc prolonging effect cannot be excluded.
Osimertinib did not cause genetic damage in in vitro and in vivo assays. Osimertinib showed no carcinogenic potential when administered orally to Tg rasH2 transgenic mice for 26 weeks. An increased incidence of proliferative vascular lesions (angiomatous hyperplasia and haemangioma) in the mesenteric lymph node was observed in the rat 104-week carcinogenicity study at exposures 0.2 times the AUC at the recommended clinical dose of 80 mg once daily, and is unlikely to be relevant for humans.
Reproductive toxicity
Degenerative changes were present in the testes in rats and dogs exposed to osimertinib for ≥1 month and there was a reduction in male fertility in rats following exposure to osimertinib for 3 months. These findings were seen at clinically relevant plasma concentrations. Pathology findings in the testes seen following 1 month dosing were reversible in rats; however, a definitive statement on reversibility of these lesions in dogs cannot be made.
Based on studies in animals, female fertility may be impaired by treatment with osimertinib. In repeat dose toxicity studies, an increased incidence of anoestrus, corpora lutea degeneration in the ovaries and epithelial thinning in the uterus and vagina were seen in rats exposed to osimertinib for ≥1 month at clinically relevant plasma concentrations. Findings in the ovaries seen following 1 month dosing were reversible. In a female fertility study in rats, administration of osimertinib at 20 mg/kg/day (approximately equal to the recommended daily clinical dose of 80 mg) had no effects on oestrus cycling or the number of females becoming pregnant, but caused early embryonic deaths. These findings showed evidence of reversibility following a 1 month off-dose.
In a modified embryofoetal development study in the rat, osimertinib caused embryolethality when administered to pregnant rats prior to embryonic implantation. These effects were seen at a maternally tolerated dose of 20 mg/kg where exposure was equivalent to the human exposure at the recommended dose of 80 mg daily (based on total AUC). Exposure at doses of 20 mg/kg and above during organogenesis caused reduced foetal weights but no adverse effects on external or visceral foetal morphology. When osimertinib was administered to pregnant female rats throughout gestation and then through early lactation, there was demonstrable exposure to osimertinib and its metabolites in suckling pups plus a reduction in pup survival and poor pup growth (at doses of 20 mg/kg and above).
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.