TALZENNA Hard capsule Ref.[7615] Active ingredients: Talazoparib
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: Pfizer Europe MA EEIG, Boulevard de la Plaine 17, 1050 Bruxelles, Belgium
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: antineoplastic agents, other antineoplastic agents
ATC code: L01XK04
Mechanism of action
Talazoparib is an inhibitor of PARP enzymes, PARP1 (IC50=0.7 nM), and PARP2 (IC50=0.3 nM). PARP enzymes are involved in cellular DNA damage response signalling pathways such as DNA repair, gene transcription, and cell death. PARP inhibitors (PARPi) exert cytotoxic effects on cancer cells by 2 mechanisms, inhibition of PARP catalytic activity and by PARP trapping, whereby PARP protein bound to a PARPi does not readily dissociate from a DNA lesion, thus preventing DNA repair, replication, and transcription, thereby resulting in apoptosis and/or cell death. Treatment of cancer cell lines that are harbouring defects in DNA repair genes with talazoparib single agent leads to increased levels of γH2AX, a marker of double stranded DNA breaks, and results in decreased cell proliferation and increased apoptosis. Talazoparib anti-tumour activity was also observed in a patient-derived xenograft (PDX) BRCA mutant breast cancer model where the patient was previously treated with a platinum-based regimen, as well as in an androgen receptor (AR)-positive prostate cancer xenograft model. In these PDX models talazoparib decreased tumour growth and increased γH2AX level and apoptosis in the tumours.
The anti-tumour effects of combined inhibition of PARP and AR activity is based on the following mechanisms: AR signalling inhibition suppresses the expression of homologous recombination repair (HRR) genes including BRCA1, resulting in sensitivity to PARP inhibition. PARP1 activity has been shown to be required for maximal AR function and thus inhibiting PARP may reduce AR signalling and increase sensitivity to AR signalling inhibitors. Clinical resistance to AR blockade is sometimes associated with co-deletion of RB1 and BRCA2, which is in turn associated with sensitivity to PARP inhibition.
Cardiac electrophysiology
The effect of talazoparib on cardiac repolarisation was evaluated using time-matched electrocardiograms (ECGs) in assessing the relationship between the change of the QT interval corrected for heart rate (QTc) from baseline and the corresponding plasma talazoparib concentrations in 37 patients with advanced solid tumours. Talazoparib did not have a clinically relevant effect on QTc prolongation at the maximum clinically recommended monotherapy dose of 1 mg once daily.
Clinical efficacy and safety
Germline BRCA-mutated (gBRCAm) HER2-negative locally advanced or metastatic breast cancer
EMBRACA study
EMBRACA was an open-label, randomised, parallel, 2-arm multicentre study of Talzenna versus chemotherapy (capecitabine, eribulin, gemcitabine, vinorelbine) in patients with germline BRCA-mutated HER2-negative locally advanced or metastatic breast cancer who received no more than 3 prior cytotoxic chemotherapy regimens for their metastatic or locally advanced disease. Patients were required to have received treatment with an anthracycline and/or a taxane (unless contraindicated) in the neoadjuvant, adjuvant and/or metastatic setting. Patients with prior platinum therapy for advanced disease were required to have no evidence of disease progression during platinum therapy. No prior treatment with a PARPi was permitted.
Of the 431 patients randomised in the EMBRACA study, 408 (95%) were centrally confirmed to have a deleterious or suspected deleterious gBRCAm using a clinical study assay; out of which 354 (82%) were confirmed using the BRACAnalysis CDx. BRCA mutation status (breast cancer susceptibility gene 1 [BRCA1] positive or breast cancer susceptibility gene 2 [BRCA2] positive) was similar across both treatment arms.
A total of 431 patients were randomised 2:1 to receive Talzenna 1 mg capsules once daily or chemotherapy at standard doses until progression or unacceptable toxicity. Of the 431 patients randomised onto EMBRACA, 287 were randomised to the Talzenna arm and 144 to the chemotherapy arm. Randomisation was stratified by prior use of chemotherapy for metastatic disease (0 versus 1, 2, or 3), by triple-negative disease status (triple-negative breast cancer [TNBC] versus non-TNBC), and history of central nervous system metastasis (yes versus no).
Patient demographic, baseline, and disease characteristics were generally similar between the study treatment arms (see Table 5).
Table 5. Demographic, baseline, and disease characteristics—EMBRACA study:
| Talazoparib (N=287) | Chemotherapy (N=144) | |
|---|---|---|
| Median age (y [range]) | 45.0 (27.0, 84.0) | 50.0 (24.0, 88.0) |
| Age category (y), n (%) | ||
| <50 | 182 (63.4%) | 67 (46.5%) |
| 50 to <65 | 78 (27.2%) | 67 (46.5%) |
| ≥65 | 27 (9.4%) | 10 (6.9%) |
| Gender, n (%) | ||
| Female | 283 (98.6%) | 141 (97.9%) |
| Male | 4 (1.4%) | 3 (2.1%) |
| Race, n (%) | ||
| Asian | 31 (10.8%) | 16 (11.1%) |
| Black or African American | 12 (4.2%) | 1 (0.7%) |
| White | 192 (66.9%) | 108 (75.0%) |
| Other | 5 (1.7%) | 1 (0.7%) |
| Not reported | 47 (16.4%) | 1 8 (12.5%) |
| ECOG performance status, n (%) | ||
| 0 | 153 (53.3%) | 84 (58.3%) |
| 1 | 127 (44.3%) | 57 (39.6%) |
| 2 | 6 (2.1%) | 2 (1.4%) |
| Missing | 1 (0.3%) | 1 (0.7%) |
| Hormone receptor status, n (%) | ||
| HER2-positive | 0 (0.0%) | 0 (0.0%) |
| Triple-negative | 130 (45.3%) | 60 (41.7%) |
| Hormone receptor-positive (ER positive or PgR positive) | 157 (54.7%) | 84 (58.3%) |
| BRCA status by central or local laboratory assessment, n (%) | 287 (100.0%) | 144 (100.0%) |
| BRCA1-mutation positive | 133 (46.3%) | 63 (43.8%) |
| BRCA2-mutation positive | 154 (53.7%) | 81 (56.3%) |
| Time from initial diagnosis of breast cancer to diagnosis of advanced breast cancer (years) | ||
| n | 286 | 144 |
| Median | 1.9 | 2.7 |
| Minimum, maximum | 0, 22 | 0, 24 |
| Categories for time from initial diagnosis of breast cancer to diagnosis of advanced breast cancer | ||
| <12 months | 108 (37.6%) | 42 (29.2%) |
| ≥12 months | 178 (62.0%) | 102 (70.8%) |
| Number of prior cytotoxic regimens for locally advanced or metastatic disease | ||
| Mean (Std Dev) | 0.9 (1.01) | 0.9 (0.89) |
| Median | 1 | 1 |
| Minimum, maximum | 0, 4 | 0, 3 |
| Number of patients who received prior cytotoxic regimens for locally advanced or metastatic disease, n (%) | ||
| 0 | 111 (38.7%) | 54 (37.5%) |
| 1 | 107 (37.3%) | 54 (37.5%) |
| 2 | 57 (19.9%) | 28 (19.4%) |
| 3 | 11 (3.8%) | 8 (5.6%) |
| ≥4 | 1 (0.3%) | 0 (0.0%) |
| Number of patients who received following prior therapies, n (%) | ||
| Taxane | 262 (91.3%) | 130 (90.3%) |
| Anthracycline | 243 (84.7%) | 115 (79.9%) |
| Platinum | 46 (16.0%) | 30 (20.8%) |
Abbreviations: BRCA=breast cancer susceptibility gene; ER=estrogen receptor; HER2=human epidermal growth factor receptor 2; N=number of patients; n=number of patients in category; PgR=progesterone receptor.
The primary efficacy endpoint was progression-free survival (PFS) evaluated according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1, as assessed by blinded independent central review (BICR). The secondary objectives were objective response rate (ORR), overall survival (OS), safety, and PK.
The study demonstrated a statistically significant improvement in PFS, the primary efficacy outcome, for Talzenna compared with chemotherapy. There was no statistically significant effect on OS at the time of final OS analysis. Efficacy data for EMBRACA are summarised in Table 6. The Kaplan-Meier curves for PFS and OS are displayed in Figure 1 and Figure 3, respectively.
Table 6. Summary of efficacy results — EMBRACA study*:
| Talazoparib | Chemotherapy | |
|---|---|---|
| PFS by BICR | N=287 | N=144 |
| Events, number (%) | 186 (65%) | 83 (58%) |
| Median (95% CI), months | 8.6 (7.2, 9.3) | 5.6 (4.2, 6.7) |
| Hazard ratioa (95% CI) | 0.54 (0.41, 0.71) | |
| 2-sided p-valueb | p<0.0001 | |
| OS (final analysis)c | N=287 | N=144 |
| Events, number (%) | 216 (75.3%) | 108 (75%) |
| Median (95% CI), months | 19.3 (16.6, 22.5) | 19.5 (17.4, 22.4) |
| Hazard ratioa (95% CI) | 0.85 (0.67, 1.07)c | |
| 2-sided p-valueb | p=0.1693 | |
| Objective response by investigatord,e | N=219 | N=114 |
| ORR, % (95% CI) | 62.6 (55.8, 69.0) | 27.2 (19.3, 36.3) |
| Odds ratio (95% CI) | 4.99 (2.93, 8.83) | |
| 2-sided p-valuef | p<0.0001 | |
| Duration of response by investigatord | N=137 | N=31 |
| Median (IQR), months | 5.4 (2.8, 11.2) | 3.1 (2.4, 6.7) |
Abbreviations: BICR=blinded independent central review; CI=confidence interval;
CMH=Cochran-Mantel-Haenszel; CR=complete response; IQR=interquartile range; ITT=intent-to-treat; N=number of patients; ORR=objective response rate; OS=overall survival; PARP=poly (adenosine diphosphate-ribose) polymerase; PFS=progression-free survival; PR=partial response; RECIST 1.1=Response Evaluation Criteria in Solid Tumors version 1.1.
* PFS, ORR and Duration of response are based on the data cutoff date of 15 September 2017 and a median follow-up for PFS of 13.0 months (95% CI: 11.1, 18.4) in the talazoparib arm and 7.2 months (95% CI: 4.6, 11.1) in the chemotherapy arm. OS is based on the data cutoff date 30 September 2019 and a median follow-up of 44.9 months (95% CI: 37.9, 47.0) in the talazoparib arm and 36.8 months (95% CI: 34.3, 43.0) in the chemotherapy arm.
a Hazard ratio was based on stratified Cox regression model with treatment as the only covariate (stratification factors: number of prior cytotoxic chemotherapy regimens, triple-negative status, history of central nervous system metastasis) and was relative to overall chemotherapy with <1 favouring talazoparib.
b Stratified log-rank test.
c At the time of the final OS analysis, 46.3% versus 41.7% of patients randomised in the talazoparib and chemotherapy arms, respectively, received subsequently a platinum therapy, and 4.5% versus 32.6% received subsequently a PARP inhibitor treatment.
d Conducted in ITT with measurable disease population who had an objective response. The complete response rate was 5.5% for talazoparib compared to 0% for the chemotherapy arm.
e Per RECIST 1.1, confirmation of CR/PR was not required.
f Stratified CMH test.
Figure 1. Kaplan-Meier curves of PFS — EMBRACA study:
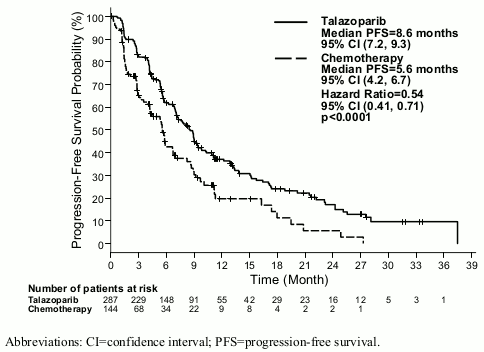
Abbreviations: CI=confidence interval; PFS=progression-free survival.
A series of prespecified subgroup PFS analyses was performed based on prognostic factors and baseline characteristics to investigate the internal consistency of treatment effect. Consistent with the overall results, a reduction in the risk of disease progression or death in favour of the talazoparib arm was observed in all individual patient subgroups (Figure 2).
Figure 2. Forest plot of PFS analyses for key subgroups — EMBRACA study:
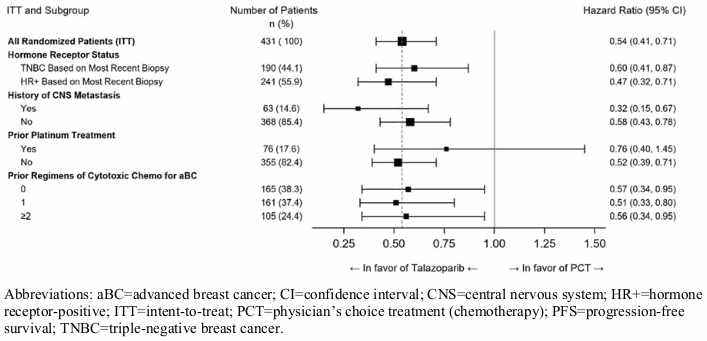
Abbreviations: aBC=advanced breast cancer; CI=confidence interval; CNS=central nervous system; HR+=hormone receptor-positive; ITT=intent-to-treat; PCT=physician's choice treatment (chemotherapy); PFS=progression-free survival; TNBC=triple-negative breast cancer.
Figure 3. Kaplan-Meier curves of overall survival — EMBRACA study:

Abbreviations: CI=confidence interval; OS=overall survival.
Primary analysis' p-value was based on a stratified log-rank test.
Metastatic castration-resistant prostate cancer (mCRPC)
TALAPRO-2 study
TALAPRO-2 was a randomised, double-blind, placebo-controlled study in which patients (N=805) with mCRPC were randomised 1:1 to receive Talzenna 0.5 mg once daily in combination with enzalutamide 160 mg once daily, versus a comparator arm of placebo in combination with enzalutamide 160 mg once daily. All patients received a gonadotropin-releasing hormone (GnRH) analog or had prior bilateral orchiectomy and needed to have progressed on prior androgen deprivation therapy. Prior treatment with abiraterone or taxane-based chemotherapy for metastatic castration-sensitive prostate cancer (mCSPC) was permitted.
Randomisation was stratified by (1) previous treatment with abiraterone or taxane-based chemotherapy versus no such prior treatment; and by (2) HRR gene mutation status which was prospectively tested by next generation sequencing of tumour tissue using FoundationOne CDx or circulating tumour DNA (ctDNA) using FoundationOne Liquid CDx; patients with tumour HRR gene mutations (ATM, ATR, BRCA1, BRCA2, CDK12, CHEK2, FANCA, MLH1, MRE11A, NBN, PALB2, or RAD51C) versus patients without tumour HRR gene mutations or with unknown status.
The median age was 71 years (range 36 to 91) in both arms; 62% were White, 31% were Asian, and 2% were Black. Most participants (66%) in both arms had an ECOG performance status of 0. In patients treated with Talzenna, the proportion of patients with RECIST 1.1 measurable disease at baseline per BICR was 30%. Twenty-eight percent (28%) of patients had received prior abiraterone or taxane-based chemotherapy. Twenty percent (20%) had tumours with HRR gene mutations and 80% had tumours that did not have HRR gene mutations or had an unknown status.
The primary efficacy outcome was radiographic progression-free survival (rPFS) evaluated according to RECIST version 1.1 and Prostate Cancer Clinical Trials Working Group Criteria 3 (PCWG3) (bone) criteria, as assessed by BICR. OS was an alpha-controlled secondary endpoint.
A statistically significant improvement in BICR-assessed rPFS was demonstrated for Talzenna in combination with enzalutamide compared to placebo in combination with enzalutamide. A sensitivity analysis of investigator-assessed rPFS was consistent with the BICR-assessed rPFS results.
Efficacy results of TALAPRO-2 are provided in Table 7 and Figure 4.
Table 7. Summary of efficacy results — TALAPRO-2 (mCRPC)*:
| Talazoparib + enzalutamide | Placebo + enzalutamide | |
|---|---|---|
| rPFS by BICR | N=402 | N=403 |
| Events, number (%) | 151 (37.6) | 191 (47.4) |
| Median months (95% CI) | NR (27.5, NR) | 21.9 (16.6, 25.1) |
| Hazard ratio (95% CI)a p-valueb | 0.627 (0.506, 0.777) p<0.0001 | |
| Second interim OS | ||
| Events, number (%) | 156 (38.8) | 174 (43.2) |
| Median months (95% CI) | NR (37.3, NR) | 38.2 (34.1, 43.1) |
| Hazard ratio (95% CI)a | 0.837 (0.674, 1.040) | |
Abbreviations: BICR=blinded independent central review; CI=confidence interval; CSPC=castration-sensitive prostate cancer; HRR=homologous recombination repair; mCRPC=metastatic castration-resistant prostate cancer; N=number of patients; NHT=novel hormone therapy; NR=not reached; OS-overall survival; rPFS=radiographic progression-free survival.
* rPFS is based on the data cutoff date of 16 August 2022 and a median follow-up for rPFS of 24.9 months (95% CI: 24.7, 25.3) in the talazoparib plus enzalutamide arm and 24.6 months (95% CI: 22.1, 24.9) in the placebo plus enzalutamide arm. Second interim OS is based on the data cutoff date 28 March 2023 and a median follow-up of 35.8 months (95% CI: 33.6, 35.9) in the talazoparib plus enzalutamide arm and 34.6 months (95% CI: 32.7, 35.9) in the placebo plus enzalutamide arm.
a Hazard ratio based on Cox proportional hazards model stratified by previous treatment with NHT (abiraterone) or taxane-based chemotherapy for CSPC (yes versus no) and by HRR mutational status (deficient versus non-deficient/unknown) with <1 favouring talazoparib.
b P-values (2-sided) from the log-rank test stratified by previous treatment with NHT (abiraterone) or taxane-based chemotherapy for CSPC and by HRR mutational status.
Table 8. Summary of efficacy results for subgroup analysis – TALAPRO-2 (mCRPC)*:
| Talazoparib + enzalutamide | Placebo + enzalutamide | |
|---|---|---|
| HRRm Subgroup Analysesa | ||
| HRRm | N=85 | N=82 |
| rPFS by BICR | ||
| Events, number (%) | 37 (43.5) | 49 (59.7) |
| Median months (95% CI) | 27.9 (16.8, NR) | 13.8 (10.9, 19.5) |
| Hazard ratio (95% CI)b | 0.424 (0.275, 0.653) | |
| Second interim OS | ||
| Events, number (%) | 30 (35.3) | 41 (50.0) |
| Median months (95% CI) | 41.9 (36.4, NR) | 30.8 (25.6, 38.8) |
| Hazard ratio (95% CI)b | 0.516 (0.320, 0.831) | |
| Non-HRRm | N=207 | N=219 |
| rPFS by BICR | ||
| Events, number (%) | 73 (35.3) | 95 (43.4) |
| Median months (95% CI) | NR (25.8, NR) | 22.4 (16.6, NR) |
| Hazard ratio (95% CI)b | 0.695 (0.511, 0.944) | |
| Second interim OS | ||
| Events, number (%) | 82 (39.6) | 96 (43.8) |
| Median months (95% CI) | NR (33, NR) | 38 (33.9, NR) |
| Hazard ratio (95% CI)b | 0.880 (0.654, 1.182) | |
| BRCAm Subgroup Analysesa | ||
| BRCAm | N=27 | N=32 |
| rPFS by BICR | ||
| Events, number (%) | 8 (29.6) | 22 (68.7) |
| Median months (95% CI) | NR (16.8, NR) | 11 (7.4, 24.6) |
| Hazard ratio (95% CI)b | 0.232 (0.101, 0.529) | |
| Second interim OS | ||
| Events, number (%) | 12 (44.4) | 18 (56.3) |
| Median months (95% CI) | 41.9 (24.9, NR) | 26.1 (15.2, NR) |
| Hazard ratio (95% CI)b | 0.558 (0.263, 1.187) | |
Abbreviations: BICR=blinded independent central review; BRCAm=breast cancer gene mutated; CI=confidence interval; CSPS=castration-sensitive prostate cancer; ctDNA=circulating tumour DNA; HRRm=homologous recombination repair gene mutated; mCRPC=metastatic castration-resistant prostate cancer; N=number of patients; NHT=novel hormone therapy; NR=not reached; OS=overall survival; rPFS=radiographic progression-free survival.
* Based on the data cutoff date of 16 August 2022 and a median follow-up for rPFS of 24.9 months (95% CI: 24.7, 25.3) in the talazoparib plus enzalutamide arm, and 24.6 months (95% CI: 22.1, 24.9) in the placebo plus enzalutamide arm. Second interim OS is based on the data cutoff date 28 March 2023 and a median follow-up of 35.8 months (95% CI: 33.6, 35.9) in the talazoparib plus enzalutamide arm and 34.6 months (95% CI: 32.7, 35.9) in the placebo plus enzalutamide arm.
a Derived based on prospective tumour tissue-based results (results known prior to randomisation) and prospective blood-based ctDNA results (results known prior to randomisation).
b Hazard ratio based on Cox proportional hazard model stratified by previous treatment with NHT (abiraterone) or taxane-based chemotherapy for CSPC (yes versus no) with <1 favouring talazoparib.
Figure 4. Kaplan-Meier curves of rPFS by BICR — TALAPRO-2 (mCRPC):

Abbreviations: BICR=blinded independent central review; CI=confidence interval; mCRPC=metastatic castration-resistant prostate cancer; PFS=progression-free survival; rPFS=radiographic progression-free survival.
Figure 5. Forest plot of rPFS analyses for key subgroups — TALAPRO-2 (mCRPC):

Abbreviations: CI=confidence interval; ctDNA=circulating tumour DNA; ENZA=enzalutamide; HRR=homologous recombination repair; HRRm=homologous recombination repair gene mutated; IWRS=Interactive Web Response System; mCRPC=metastatic castration-resistant prostate cancer; N=number of participants; NE=not evaluable/not reached; NHT=novel hormone therapy; PBO=placebo; PSA=prostate-specific antigen; rPFS=radiographic progression-free survival; SE=study entry; TALA=talazoparib; w/o=without.
Hazard ratio for all patients was based on a Cox model stratified by the randomization stratification factors. For all subgroups, hazard ratio was based on an unstratified Cox model with treatment as the only covariate. A hazard ratio <1 favours talazoparib.
HRR status is derived based on prospective tumour tissue-based results and prospective blood-based ctDNA results.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with talazoparib in all subsets of the paediatric population in breast cancer and prostate cancer (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Talazoparib exposure generally increased proportionally with dose across the range of 0.025 mg to 2 mg after daily administration of multiple doses. Following repeated daily dosing of 1 mg talazoparib monotherapy to breast cancer patients, the geometric mean (% coefficient of variation [CV%]) area under the plasma concentration-time curve (AUC) and maximum observed plasma concentration (Cmax) of talazoparib at steady-state was in the range of 126 (107) ng•hr/mL to 208 (37) ng•hr/mL and 11 (90) ng/mL to 19 (27) ng/mL, respectively. After oral administration of 0.5 mg talazoparib once daily in combination with enzalutamide in patients with mCRPC, the geometric mean (CV%) steady-state Ctrough across visits ranged from 3.29 to 3.68 ng/mL (45 to 48%), which is similar to the observed values of 3.53 (61%) ng/mL when talazoparib monotherapy was administered at 1 mg once daily in breast cancer patients. Following repeated daily dosing, talazoparib plasma concentrations reached steady-state within 2 to 3 weeks when administered alone, and approximately within 9 weeks when co-administered with enzalutamide. The median accumulation ratio of talazoparib following repeated oral administration of 1 mg monotherapy once daily was in the range of 2.3 to 5.2. Talazoparib is a substrate of P-gp and BCRP transporters.
Absorption
Following oral administration of talazoparib, the median time to Cmax (Tmax) was generally between 1 to 2 hours after dosing. The absolute bioavailability study has not been conducted in humans. However, based on urinary excretion data the absolute bioavailability is at least 41% with fraction absorbed of at least 69% (see Elimination). No significant effect of acid-reducing agents on talazoparib exposure is expected, given sufficient solubility of talazoparib at all pHs between 1 and 6.8. Twenty-eight percent (28%) of the patients in the pivotal study were taking acid-reducing agents, mainly proton pump inhibitors.
The effect of food
Food intake decreased the rate but not the extent of talazoparib absorption. Following a single oral dose of talazoparib with high-fat, high-calorie food (approximately 827 calories, 57% fat), the mean Cmax of talazoparib was decreased by approximately 46%, the median Tmax was delayed from 1 to 4 hours, while the AUCinf was not affected. Based on these results, Talzenna can be administered with or without food (see section 4.2).
Distribution
The population mean apparent volume of distribution (Vss/F) of talazoparib was 420 L. In vitro, talazoparib is approximately 74% bound to plasma proteins with no concentration dependence over the concentration range of 0.01 μM to 1 μM. Renal or hepatic impairment does not appear to impact talazoparib protein binding as there was no obvious trend in the mean talazoparib fraction of unbound drug (fu) in human plasma in vivo with worsening renal function or hepatic function.
Biotransformation
Talazoparib undergoes minimal hepatic metabolism in humans. Following oral administration of a single 1 mg dose of [14C]talazoparib to humans, no major circulating metabolites were identified in plasma, and talazoparib was the only circulating drug-derived entity identified. No metabolites that individually represented more than 10% of the administered dose were recovered in the urine or faeces.
In vitro, talazoparib was not an inhibitor of cytochrome (CYP)1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, or CYP3A4/5 or inducer of CYP1A2, CYP2B6, or CYP3A4 at clinically relevant concentrations.
In vitro, talazoparib did not inhibit any of the major intestinal, hepatic or renal membrane transporters (P-gp, BCRP, organic anion transporting polypeptide [OATP]1B1, OATP1B3, organic cationic transporter [OCT]1, OCT2, organic anion transporter [OAT]1, OAT3, bile salt export pump [BSEP], multidrug and toxin extrusion [MATE]1 and MATE2-K) at clinically relevant concentrations.
In vitro, talazoparib did not inhibit any of the major uridine-diphosphate glucuronosyltransferase (UGT) isoforms (1A1, 1A4, 1A6, 1A9, 2B7, and 2B15) at clinically relevant concentrations.
Elimination
Renal elimination of unchanged drug (passive filtration and active secretion) is the major route of talazoparib elimination. P-gp is likely involved in talazoparib active renal secretion. The mean (±standard deviation) terminal plasma half-life of talazoparib was 90 (±58) hours and the population mean (inter-subject variability) apparent oral clearance (CL/F) was 6.5 (31%) L/h in cancer patients. In 6 female patients given a single oral dose of [14C]talazoparib, a mean of 69% (±8.6%) and 20% (±5.5%) of the total administered radioactive dose was recovered in urine and faeces, respectively. Excretion of unchanged talazoparib in urine was the major route of elimination accounting for 55% of the administered dose, while unchanged talazoparib recovered in the faeces accounted for 14%.
Special populations
Age, sex, and body weight
A population PK analysis was conducted using data from 490 patients with cancer who received talazoparib 1 mg daily as monotherapy to evaluate the impact of age (ranging from 18 to 88 years), sex (53 males and 437 females), and body weight (ranging from 35.7 kg to 162 kg) on the PK of talazoparib. The results have shown that age, sex, and body weight had no clinically relevant effect on the PK of talazoparib.
Race
Based on a population PK analysis that included 490 patients who received talazoparib 1 mg daily as monotherapy, where 41 patients were Asian and 449 patients were Non-Asian (361 White, 16 Black, 9 Others, and 63 Not reported), talazoparib CL/F was higher in Asian patients compared to Non-Asian patients, leading to 19% lower exposure (AUC) in Asian patients.
Paediatric population
Pharmacokinetics of talazoparib have not been evaluated in patients < 18 years of age.
Renal impairment
Talazoparib monotherapy:
Data from a PK study in advanced cancer patients with varying degrees of renal impairment indicated that talazoparib total exposure (AUC0-24) after multiple talazoparib once daily doses increased by 92% and 169% in patients with moderate (eGFR 30 – < 60 mL/min) and severe (eGFR < 30 mL/min) renal impairment, respectively, relative to patients with normal renal function (eGFR ≥90 mL/min).
Talazoparib Cmax increased by 90% and 107% in patients with moderate and severe renal impairment, respectively, relative to patients with normal renal function. Talazoparib exposure was similar for patients with mild renal impairment (eGFR 60 – < 90 mL/min) and those with normal renal function. In addition, based on a population PK analysis that included 490 patients, where 132 patients had mild renal impairment (60 mL/min ≤ CrCL < 90 mL/min), 33 patients had moderate renal impairment (30 mL/min ≤ CrCL < 60 mL/min), and 1 patient had severe renal impairment (CrCL < 30 mL/min),talazoparib CL/F was decreased by 14% and 37% in patients with mild and moderate renal impairment, corresponding to 17% and 59% increase in AUC, respectively, when compared to patients with normal renal function (CrCL ≥ 90 mL/min). The PK of talazoparib have not been studied in patients requiring haemodialysis (see section 4.2).
Talazoparib co-administered with enzalutamide:
Based on a population PK analysis that included 412 mCRPC patients who received talazoparib co-administered with enzalutamide, where 152 patients had mild renal impairment (60 mL/min ≤ CrCL < 90 mL/min), 72 patients had moderate renal impairment (30 mL/min ≤ CrCL < 60 mL/min), and 2 patients had severe renal impairment (CrCL < 30 mL/min), talazoparib CL/F was decreased by 8% and 27%, corresponding to increases of 9% and 37% in AUC, in patients with mild and moderate renal impairment respectively, compared to patients with normal renal function. The PK of talazoparib has not been studied in patients requiring haemodialysis (see section 4.2).
Hepatic impairment
Talazoparib monotherapy:
Based on a population PK analysis that included 490 patients who received talazoparib 1 mg daily as monotherapy, where 118 patients had mild hepatic impairment (total bilirubin ≤ 1.0 × ULN and AST > ULN, or total bilirubin > 1.0 to 1.5 × ULN and any AST), mild hepatic impairment had no effect on the PK of talazoparib. The PK of talazoparib in patients with normal hepatic function, mild hepatic impairment, moderate hepatic impairment (total bilirubin > 1.5 to 3.0 × ULN and any AST) or severe hepatic impairment (total bilirubin > 3.0 × ULN and any AST) was studied in a PK study. Population PK analysis using data from this PK study indicated that mild, moderate or severe hepatic impairment had no significant impact on the PK of talazoparib (see section 4.2).
Talazoparib co-administered with enzalutamide:
The PK of talazoparib in combination with enzalutamide has not been studied in patients with hepatic impairment (see section 4.2).
5.3. Preclinical safety data
Carcinogenicity
Carcinogenicity studies have not been conducted with talazoparib.
Genotoxicity
Talazoparib was not mutagenic in a bacterial reverse mutation (Ames) test. Talazoparib was clastogenic in an in vitro chromosomal aberration assay in human peripheral blood lymphocytes and in an in vivo micronucleus assay in rats at exposures similar to clinically relevant doses. This clastogenicity is consistent with genomic instability resulting from the primary pharmacology of talazoparib, indicating the potential for genotoxicity in humans.
Repeat-dose toxicity
In repeat-dose toxicity studies in rats and in dogs, the main findings at subtherapeutic exposures included bone marrow hypocellularity with dose-dependent decrease in haematopoietic cells, depletion of lymphoid tissue in multiple organs and atrophy and/or degenerative changes in testes, epididymis and seminiferous tubules. Additional findings at higher exposures included dose-dependent increase in apoptosis/necrosis in the gastrointestinal (GI) tract, liver and ovary. Most of the histopathologic findings were generally reversible while the testes findings were partially reversible after 4 weeks of dosing cessation. These toxicity findings are consistent with the pharmacology of talazoparib and its tissue distribution pattern.
Developmental toxicology
In an embryo-foetal development study in rats, talazoparib resulted in embryo-foetal death, foetal malformation (depressed eye bulge, small eye, split sternebrae, fused cervical vertebral arch) and structural variations in bones at a maternal systemic AUC24 exposure approximately 0.09-fold the relevant human exposure at the recommended dose.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.