Source: European Medicines Agency (EU) Revision Year: 2019 Publisher: MYLAN S.A.S, 117 Allée des Parcs, 69800 Saint-Priest, France
Hypersensitivity to the active substance or to any of the excipients listed in section 6.1.
HIV antibody testing should be offered to all HBV infected patients before initiating tenofovir disoproxil therapy (see below Co-infection with HIV-1 and hepatitis B).
While effective viral suppression with antiretroviral therapy has been proven to substantially reduce the risk of sexual transmission, a residual risk cannot be excluded. Precautions to prevent transmission should be taken in accordance with national guidelines.
Patients must be advised that tenofovir disoproxil has not been proven to prevent the risk of transmission of HBV to others through sexual contact or contamination with blood. Appropriate precautions must continue to be used.
There have been reports of a high rate of virological failure and of emergence of resistance at an early stage in HIV patients when tenofovir disoproxil was combined with lamivudine and abacavir as well as with lamivudine and didanosine as a once-daily regimen.
Tenofovir is principally eliminated via the kidney. Renal failure, renal impairment, elevated creatinine, hypophosphataemia and proximal tubulopathy (including Fanconi syndrome) have been reported with the use of tenofovir disoproxil in clinical practice (see section 4.8).
It is recommended that creatinine clearance is calculated in all patients prior to initiating therapy with tenofovir disoproxil and renal function (creatinine clearance and serum phosphate) is also monitored after two to four weeks of treatment, after three months of treatment and every three to six months thereafter in patients without renal risk factors. In patients at risk for renal impairment, a more frequent monitoring of renal function is required.
If serum phosphate is <1.5 mg/dl (0.48 mmol/l) or creatinine clearance is decreased to <50 ml/min in any adult patient receiving tenofovir disoproxil, renal function should be re-evaluated within one week, including measurements of blood glucose, blood potassium and urine glucose concentrations (see section 4.8, proximal tubulopathy). Consideration should also be given to interrupting treatment with tenofovir disoproxil in adult patients with creatinine clearance decreased to <50 ml/min or decreases in serum phosphate to <1.0 mg/dl (0.32 mmol/l). Interrupting treatment with tenofovir disoproxil should also be considered in case of progressive decline of renal function when no other cause has been identified.
Use of tenofovir disoproxil should be avoided with concurrent or recent use of a nephrotoxic medicinal product (e.g. aminoglycosides, amphotericin B, foscarnet, ganciclovir, pentamidine, vancomycin, cidofovir or interleukin-2). If concomitant use of tenofovir disoproxil and nephrotoxic agents is unavoidable, renal function should be monitored weekly.
Cases of acute renal failure after initiation of high dose or multiple non-steroidal anti-inflammatory drugs (NSAIDs) have been reported in patients treated with tenofovir disoproxil and with risk factors for renal dysfunction. If tenofovir disoproxil is co-administered with an NSAID, renal function should be monitored adequately.
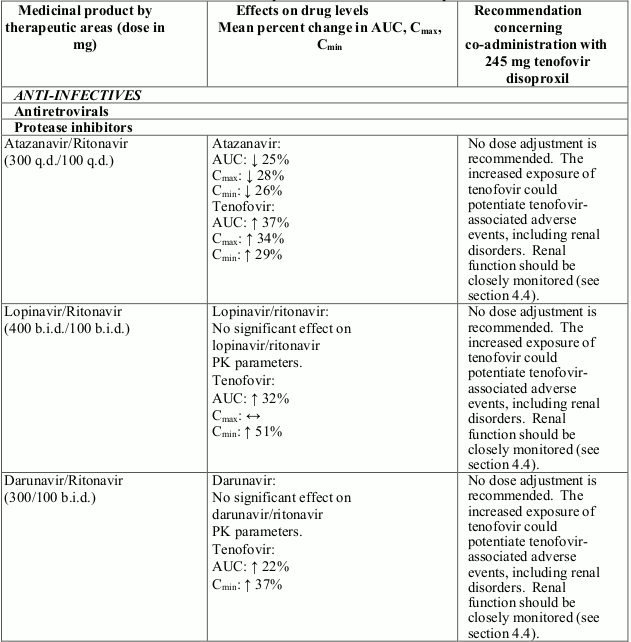
A higher risk of renal impairment has been reported in patients receiving tenofovir disoproxil in combination with a ritonavir or cobicistat boosted protease inhibitor. A close monitoring of renal function is required in these patients (see section 4.5). In patients with renal risk factors, the co- administration of tenofovir disoproxil with a boosted protease inhibitor should be carefully evaluated.
Tenofovir disoproxil has not been clinically evaluated in patients receiving medicinal products which are secreted by the same renal pathway, including the transport proteins human organic anion transporter (hOAT) 1 and 3 or MRP 4 (e.g. cidofovir, a known nephrotoxic medicinal product). These renal transport proteins may be responsible for tubular secretion and in part, renal elimination of tenofovir and cidofovir. Consequently, the pharmacokinetics of these medicinal products, which are secreted by the same renal pathway including transport proteins hOAT 1 and 3 or MRP 4, might be modified if they are co-administered. Unless clearly necessary, concomitant use of these medicinal products which are secreted by the same renal pathway is not recommended, but if such use is unavoidable, renal function should be monitored weekly (see section 4.5).
Renal safety with tenofovir disoproxil has only been studied to a very limited degree in adult patients with impaired renal function (creatinine clearance <80 ml/min).
Adult patients with creatinine clearance <50 ml/min, including haemodialysis patients:
There are limited data on the safety and efficacy of tenofovir disoproxil in patients with impaired renal function. Therefore, tenofovir disoproxil should only be used if the potential benefits of treatment are considered to outweigh the potential risks. In patients with severe renal impairment (creatinine clearance <30 ml/min) and in patients who require haemodialysis use of tenofovir disoproxil is not recommended. If no alternative treatment is available, the dosing interval must be adjusted and renal function should be closely monitored (see sections 4.2 and 5.2).
In HIV infected patients, in a 144-week controlled clinical study that compared tenofovir disoproxil with stavudine in combination with lamivudine and efavirenz in antiretroviral-naïve adult patients, small decreases in bone mineral density (BMD) of the hip and spine were observed in both treatment groups. Decreases in BMD of spine and changes in bone biomarkers from baseline were significantly greater in the tenofovir disoproxil treatment group at 144 weeks. Decreases in BMD of hip were significantly greater in this group until 96 weeks. However, there was no increased risk of fractures or evidence for clinically relevant bone abnormalities over 144 weeks.
In other studies (prospective and cross-sectional), the most pronounced decreases in BMD were seen in patients treated with tenofovir disoproxil as part of a regimen containing a boosted protease inhibitor. Alternative treatment regimens should be considered for patients with osteoporosis that are at a high risk for fractures.
Bone abnormalities (infrequently contributing to fractures) may be associated with proximal renal tubulopathy (see section 4.8).
If bone abnormalities are suspected or detected then appropriate consultation should be obtained.
There are uncertainties associated with the long term effects of bone and renal toxicity. Moreover, the reversibility of renal toxicity cannot be fully ascertained. Therefore, a multidisciplinary approach is recommended to adequately weigh on a case by case basis the benefit/risk balance of treatment, decide the appropriate monitoring during treatment (including decision for treatment withdrawal) and consider the need for supplementation.
Renal adverse reactions consistent with proximal renal tubulopathy have been reported in HIV-1 infected paediatric patients aged 2 to <12 years in clinical study GS-US-104-0352 (see sections 4.8 and 5.1).
Renal function (creatinine clearance and serum phosphate) should be evaluated prior to treatment, and monitored during treatment as in adults (see above).
If serum phosphate is confirmed to be <3.0 mg/dl (0.96 mmol/l) in any paediatric patient receiving tenofovir disoproxil, renal function should be re-evaluated within one week, including measurements of blood glucose, blood potassium and urine glucose concentrations (see section 4.8, proximal tubulopathy). If renal abnormalities are suspected or detected then consultation with a nephrologist should be obtained to consider interruption of tenofovir disoproxil treatment. Interrupting treatment with tenofovir disoproxil should also be considered in case of progressive decline of renal function when no other cause has been identified.
The same recommendations apply as in adults (see above).
The use of tenofovir disoproxil is not recommended in paediatric patients with renal impairment (see section 4.2). Tenofovir disoproxil should not be initiated in paediatric patients with renal impairment and should be discontinued in paediatric patients who develop renal impairment during tenofovir disoproxil therapy.
Tenofovir disoproxil may cause a reduction in BMD. The effects of tenofovir disoproxil-associated changes in BMD on long-term bone health and future fracture risk are currently unknown (see section 5.1).
If bone abnormalities are detected or suspected in paediatric patients, consultation with an endocrinologist and/or nephrologist should be obtained.
Safety and efficacy data are very limited in liver transplant patients.
There are limited data on the safety and efficacy of tenofovir disoproxil in HBV infected patients with decompensated liver disease and who have a Child-Pugh-Turcotte (CPT) score >9. These patients may be at higher risk of experiencing serious hepatic or renal adverse reactions. Therefore, hepatobiliary and renal parameters should be closely monitored in this patient population.
Flares on treatment: Spontaneous exacerbations in chronic hepatitis B are relatively common and are characterised by transient increases in serum ALT. After initiating antiviral therapy, serum ALT may increase in some patients (see section 4.8). In patients with compensated liver disease, these increases in serum ALT are generally not accompanied by an increase in serum bilirubin concentrations or hepatic decompensation. Patients with cirrhosis may be at a higher risk for hepatic decompensation following hepatitis exacerbation, and therefore should be monitored closely during therapy.
Flares after treatment discontinuation: Acute exacerbation of hepatitis has also been reported in patients who have discontinued hepatitis B therapy. Post-treatment exacerbations are usually associated with rising HBV DNA, and the majority appears to be self-limited. However, severe exacerbations, including fatalities, have been reported. Hepatic function should be monitored at repeated intervals with both clinical and laboratory follow-up for at least 6 months after discontinuation of hepatitis B therapy. If appropriate, resumption of hepatitis B therapy may be warranted. In patients with advanced liver disease or cirrhosis, treatment discontinuation is not recommended since post-treatment exacerbation of hepatitis may lead to hepatic decompensation.
Liver flares are especially serious, and sometimes fatal in patients with decompensated liver disease.
Co-infection with hepatitis C or D: There are no data on the efficacy of tenofovir in patients co-infected with hepatitis C or D virus.
Co-infection with HIV-1 and hepatitis B: Due to the risk of development of HIV resistance, tenofovir disoproxil should only be used as part of an appropriate antiretroviral combination regimen in HIV/HBV co-infected patients. Patients with pre-existing liver dysfunction, including chronic active hepatitis, have an increased frequency of liver function abnormalities during combination antiretroviral therapy (CART) and should be monitored according to standard practice. If there is evidence of worsening liver disease in such patients, interruption or discontinuation of treatment must be considered. However, it should be noted that increases of ALT can be part of HBV clearance during therapy with tenofovir, see above Exacerbations of hepatitis.
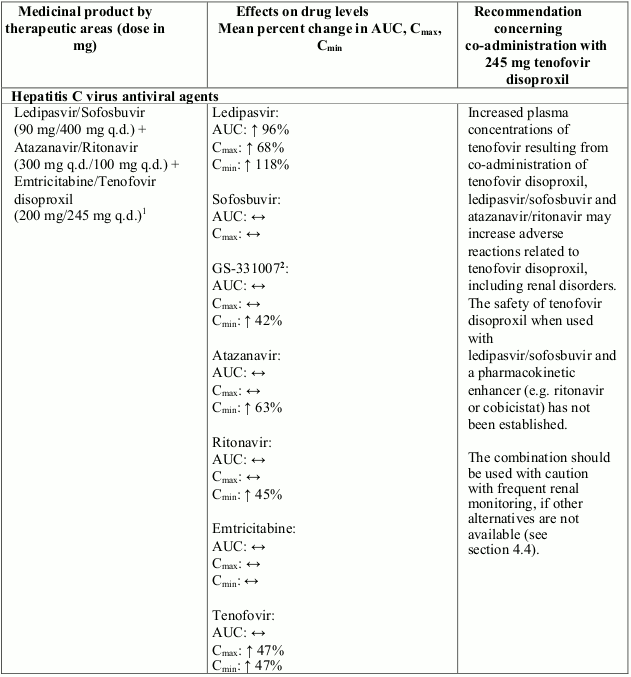
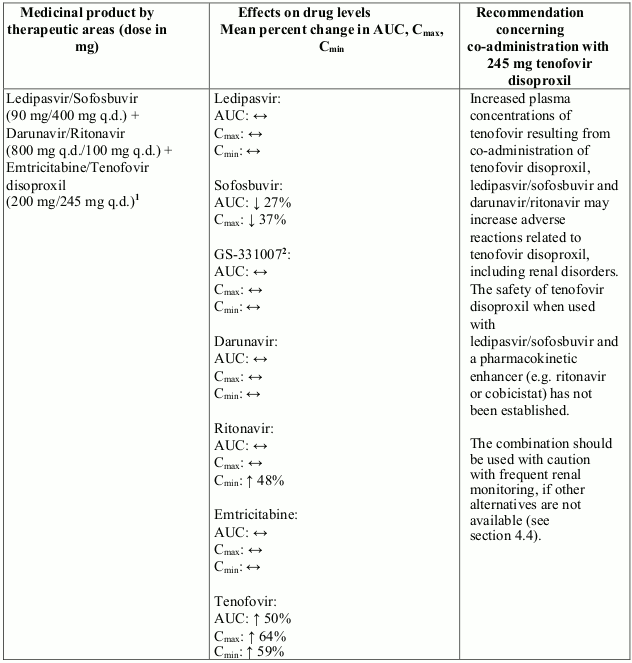
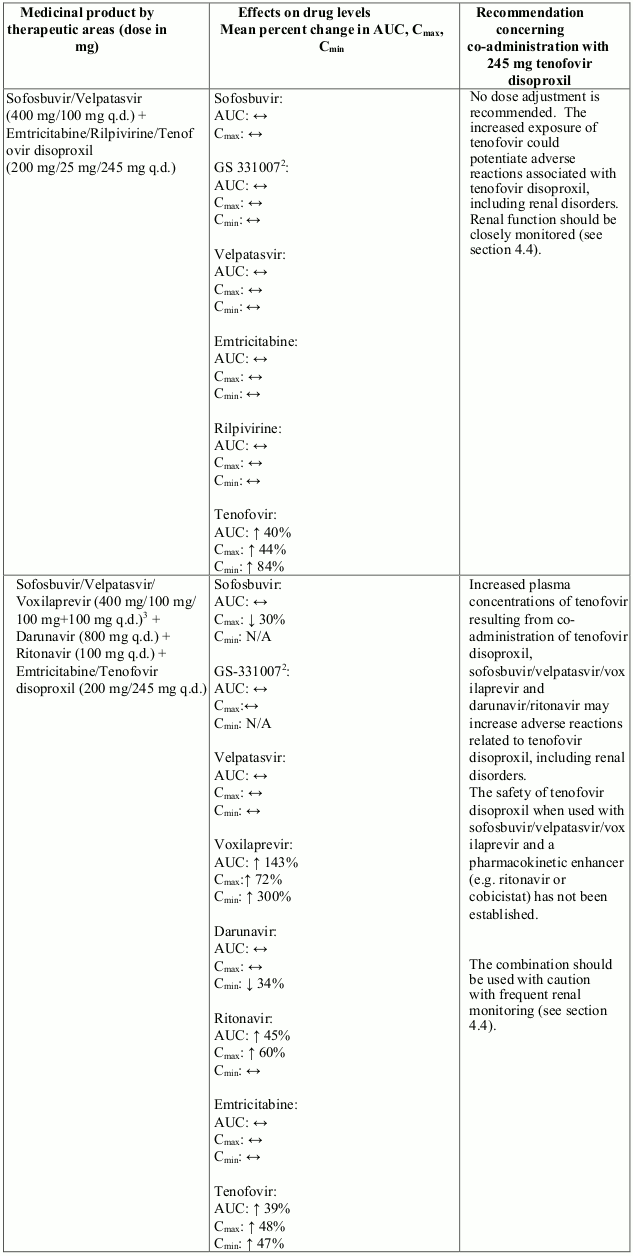
Co-administration of tenofovir disoproxil with ledipasvir/sofosbuvir, sofosbuvir/velpatasvir or sofosbuvir/velpatasvir/voxilaprevir has been shown to increase plasma concentrations of tenofovir, especially when used together with an HIV regimen containing tenofovir disoproxil and a pharmacokinetic enhancer (ritonavir or cobicistat). The safety of tenofovir disoproxil in the setting of ledipasvir/sofosbuvir, sofosbuvir/velpatasvir or sofosbuvir/velpatasvir/voxilaprevir and a pharmacokinetic enhancer has not been established. The potential risks and benefits associated with co-administration of ledipasvir/sofosbuvir, sofosbuvir/velpatasvir or sofosbuvir/velpatasvir/voxilaprevir with tenofovir disoproxil given in conjunction with a boosted HIV protease inhibitor (e.g. atazanavir or darunavir) should be considered, particularly in patients at increased risk of renal dysfunction. Patients receiving ledipasvir/sofosbuvir, sofosbuvir/velpatasvir or sofosbuvir/velpatasvir/voxilaprevir concomitantly with tenofovir disoproxil and a boosted HIV protease inhibitor should be monitored for adverse reactions related to tenofovir disoproxil.
An increase in weight and in levels of blood lipids and glucose may occur during antiretroviral therapy. Such changes may in part be linked to disease control and life style. For lipids, there is in some cases evidence for a treatment effect, while for weight gain there is no strong evidence relating this to any particular treatment. For monitoring of blood lipids and glucose reference is made to established HIV treatment guidelines. Lipid disorders should be managed as clinically appropriate.
Nucleos(t)ide analogues may impact mitochondrial function to a variable degree, which is most pronounced with stavudine, didanosine and zidovudine. There have been reports of mitochondrial dysfunction in HIV negative infants exposed in utero and/or postnatally to nucleoside analogues; these have predominantly concerned treatment with regimens containing zidovudine. The main adverse reactions reported are haematological disorders (anaemia, neutropenia) and metabolic disorders (hyperlactataemia, hyperlipasaemia). These events have often been transitory. Late onset neurological disorders have been reported rarely (hypertonia, convulsion, abnormal behaviour). Whether such neurological disorders are transient or permanent is currently unknown. These findings should be considered for any child exposed in utero to nucleos(t)ide analogues, who present with severe clinical findings of unknown etiology, particularly neurologic findings. These findings do not affect current national recommendations to use antiretroviral therapy in pregnant women to prevent vertical transmission of HIV.
In HIV infected patients with severe immune deficiency at the time of institution of CART, an inflammatory reaction to asymptomatic or residual opportunistic pathogens may arise and cause serious clinical conditions, or aggravation of symptoms. Typically, such reactions have been observed within the first few weeks or months of initiation of CART. Relevant examples are cytomegalovirus retinitis, generalised and/or focal mycobacterial infections, and Pneumocystis jirovecii pneumonia. Any inflammatory symptoms should be evaluated and treatment instituted when necessary.
Autoimmune disorders (such as Graves' disease and autoimmune hepatitis) have also been reported to occur in the setting of immune reactivation; however, the reported time to onset is more variable and these events can occur many months after initiation of treatment.
Although the aetiology is considered to be multifactorial (including corticosteroid use, alcohol consumption, severe immunosuppression, higher body mass index), cases of osteonecrosis have been reported, particularly in patients with advanced HIV disease and/or long-term exposure to CART. Patients should be advised to seek medical advice if they experience joint aches and pain, joint stiffness or difficulty in movement.
Tenofovir disoproxil has not been studied in patients over the age of 65. Elderly patients are more likely to have decreased renal function; therefore caution should be exercised when treating elderly patients with tenofovir disoproxil.
Tenofovir disoproxil Mylan 245 mg film-coated tablets contain lactose monohydrate. Consequently, patients with rare hereditary problems of galactose intolerance, total lactase deficiency, or glucose-galactose malabsorption should not take this medicinal product.
Interaction studies have only been performed in adults.
Based on the results of in vitro experiments and the known elimination pathway of tenofovir, the potential for CYP450-mediated interactions involving tenofovir with other medicinal products is low.
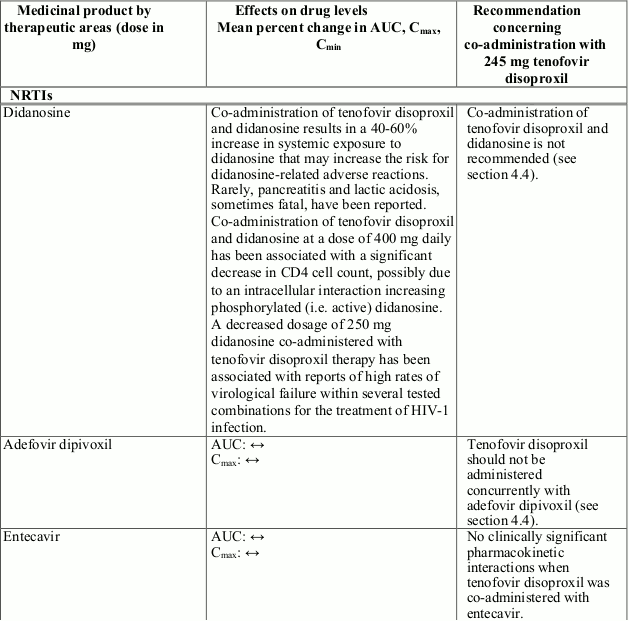
Tenofovir disoproxil should not be administered concomitantly with other medicinal products containing tenofovir disoproxil or tenofovir alafenamide.
Tenofovir disoproxil should not be administered concomitantly with adefovir dipivoxil.
Co-administration of tenofovir disoproxil and didanosine is not recommended (see section 4.4 and Table 1).
Since tenofovir is primarily eliminated by the kidneys, co-administration of tenofovir disoproxil with medicinal products that reduce renal function or compete for active tubular secretion via transport proteins hOAT 1, hOAT 3 or MRP 4 (e.g. cidofovir) may increase serum concentrations of tenofovir and/or the co-administered medicinal products.
Use of tenofovir disoproxil should be avoided with concurrent or recent use of a nephrotoxic medicinal product. Some examples include, but are not limited to, aminoglycosides, amphotericin B, foscarnet, ganciclovir, pentamidine, vancomycin, cidofovir or interleukin-2 (see section 4.4).
Given that tacrolimus can affect renal function, close monitoring is recommended when it is co-administered with tenofovir disoproxil.
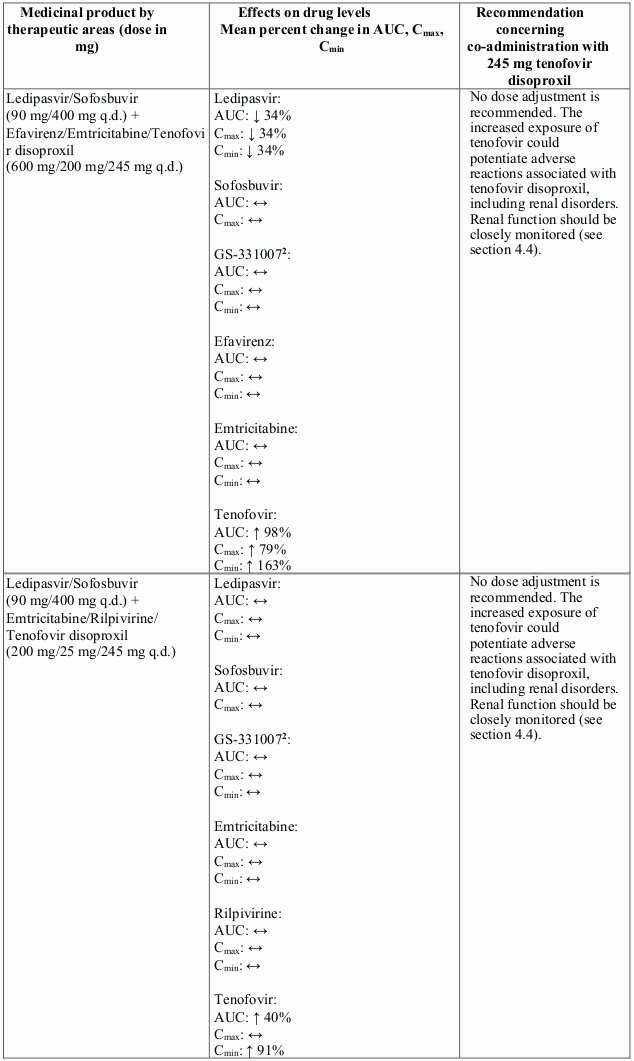
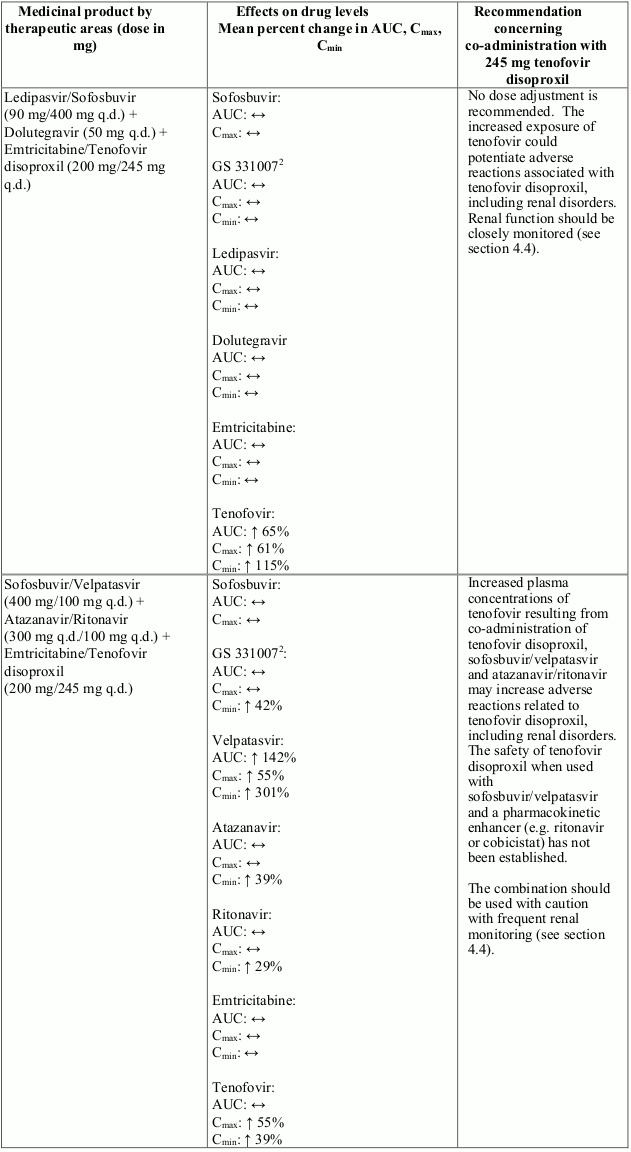
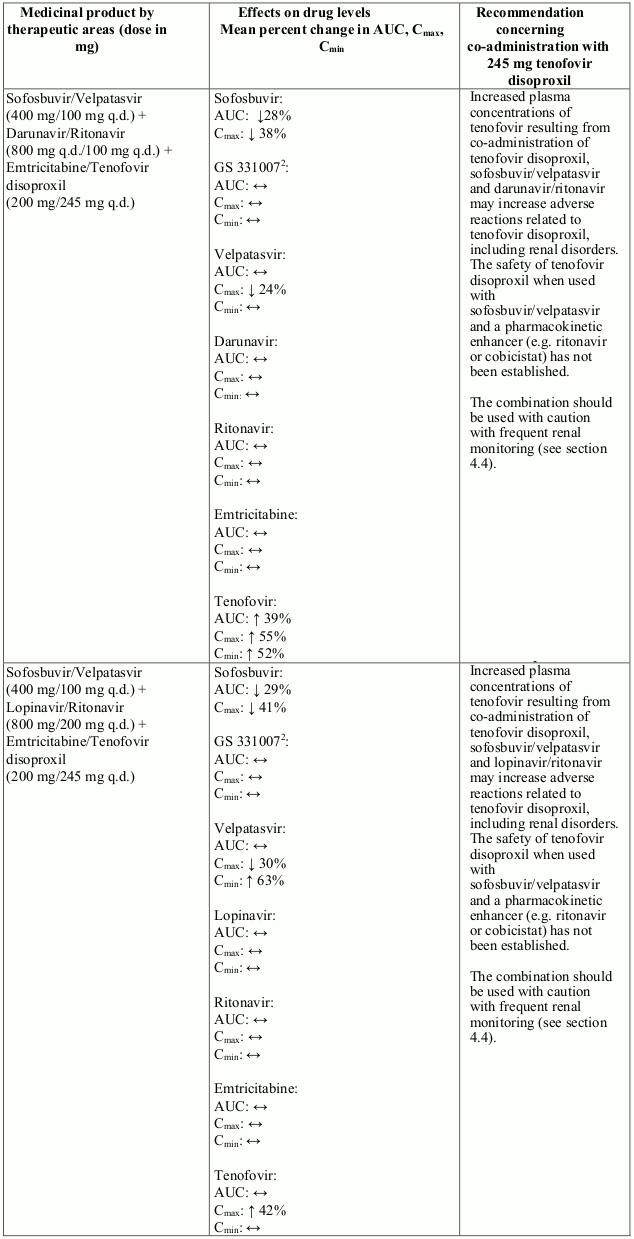
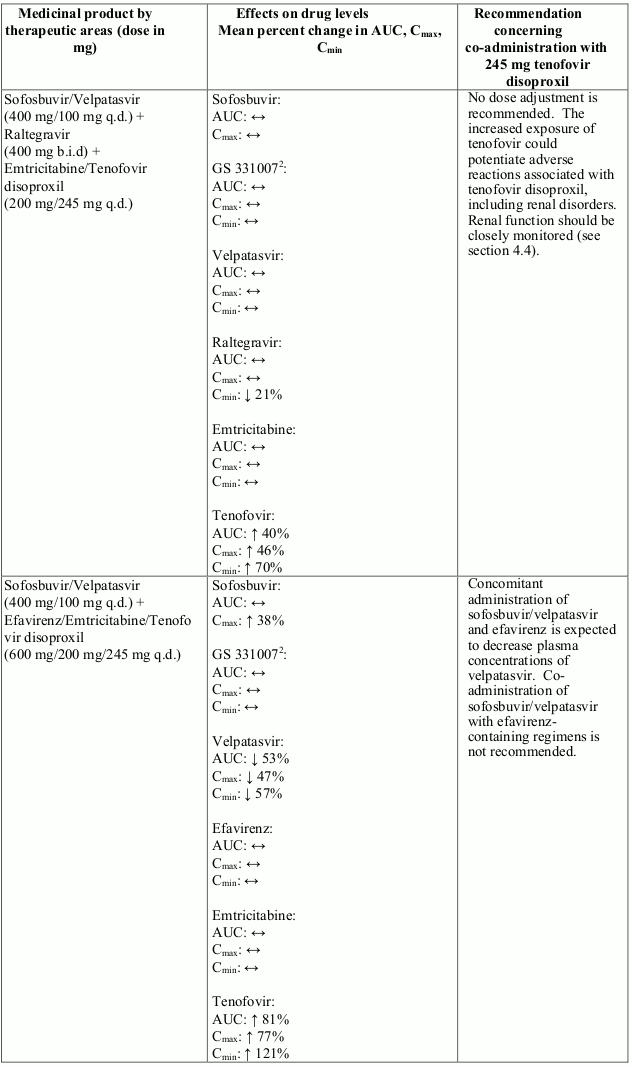
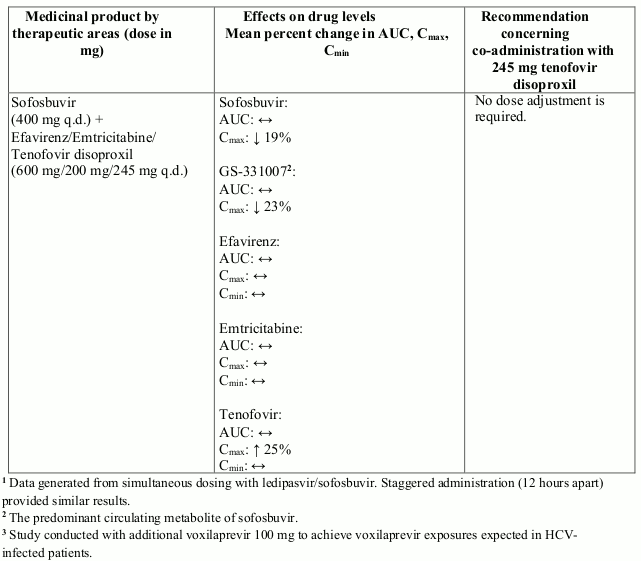
Interactions between tenofovir disoproxil and other medicinal products are listed in Table 1 below (increase is indicated as “↑”, decrease as “↓”, no change as “↔”, twice daily as “b.i.d.”, and once daily as “q.d.”).
Table 1. Interactions between tenofovir disoproxil and other medicinal products:
There were no clinically significant pharmacokinetic interactions when tenofovir disoproxil was co-administered with emtricitabine, lamivudine, indinavir, efavirenz, nelfinavir, saquinavir (ritonavir boosted), methadone, ribavirin, rifampicin, tacrolimus, or the hormonal contraceptive norgestimate/ethinyl oestradiol.
Tenofovir disoproxil must be taken with food, as food enhances the bioavailability of tenofovir (see section 5.2).
A large amount of data on pregnant women (more than 1,000 pregnancy outcomes) indicate no malformations or foetal/neonatal toxicity associated with tenofovir disoproxil. Animal studies do not indicate reproductive toxicity (see section 5.3). The use of tenofovir disoproxil may be considered during pregnancy, if necessary.
Tenofovir has been shown to be excreted in human milk. There is insufficient information on the effects of tenofovir in newborns/infants. Therefore Tenofovir disoproxil Mylan should not be used during breast-feeding.
As a general rule, it is recommended that HIV and HBV infected women do not breast-feed their infants in order to avoid transmission of HIV and HBV to the infant.
There are limited clinical data with respect to the effect of tenofovir disoproxil on fertility. Animal studies do not indicate harmful effects of tenofovir disoproxil on fertility.
No studies on the effects on the ability to drive and use machines have been performed. However, patients should be informed that dizziness has been reported during treatment with tenofovir disoproxil.
In patients receiving tenofovir disoproxil, rare events of renal impairment, renal failure and uncommon events of proximal renal tubulopathy (including Fanconi syndrome) sometimes leading to bone abnormalities (infrequently contributing to fractures) have been reported. Monitoring of renal function is recommended for patients receiving tenofovir disoproxil (see section 4.4).
Approximately one third of patients can be expected to experience adverse reactions following treatment with tenofovir disoproxil in combination with other antiretroviral agents. These reactions are usually mild to moderate gastrointestinal events. Approximately 1% of tenofovir disoproxil -treated adult patients discontinued treatment due to the gastrointestinal events.
Co-administration of tenofovir disoproxil and didanosine is not recommended as this may result in an increased risk of adverse reactions (see section 4.5). Rarely, pancreatitis and lactic acidosis, sometimes fatal, have been reported (see section 4.4).
Approximately one quarter of patients can be expected to experience adverse reactions following treatment with tenofovir disoproxil, most of which are mild. In clinical trials of HBV infected patients, the most frequently occurring adverse reaction to tenofovir disoproxil was nausea (5.4%).
Acute exacerbation of hepatitis has been reported in patients on treatment as well as in patients who have discontinued hepatitis B therapy (see section 4.4).
Assessment of adverse reactions for tenofovir disoproxil is based on safety data from clinical studies and post-marketing experience. All adverse reactions are presented in Table 2.
Assessment of adverse reactions from HIV-1 clinical study data is based on experience in two studies in 653 treatment-experienced patients receiving treatment with tenofovir disoproxil (n=443) or placebo (n=210) in combination with other antiretroviral medicinal products for 24 weeks and also in a double-blind comparative controlled study in which 600 treatment-naïve patients received treatment with tenofovir disoproxil 245 mg (n=299) or stavudine (n=301) in combination with lamivudine and efavirenz for 144 weeks.
Assessment of adverse reactions from HBV clinical study data is primarily based on experience in two double-blind comparative controlled studies in which 641 adult patients with chronic hepatitis B and compensated liver disease received treatment with tenofovir disoproxil 245 mg daily (n=426) or adefovir dipivoxil 10 mg daily (n=215) for 48 weeks. The adverse reactions observed with continued treatment for 384 weeks were consistent with the safety profile of tenofovir disoproxil. After an initial decline of approximately -4.9 ml/min (using Cockcroft-Gault equation) or -3.9 ml/min/1.73 m² (using modification of diet in renal disease [MDRD] equation) after the first 4 weeks of treatment, the rate of annual decline post baseline of renal function reported in tenofovir disoproxil treated patients was -1.41 ml/min per year (using Cockcroft-Gault equation) and -0.74 ml/min/1.73 m² per year (using MDRD equation).
The safety profile of tenofovir disoproxil in patients with decompensated liver disease was assessed in a double-blind active controlled study (GS-US-174-0108) in which adult patients received treatment with tenofovir disoproxil (n=45) or emtricitabine plus tenofovir disoproxil (n=45) or entecavir (n=22) for 48 weeks.
In the tenofovir disoproxil treatment arm, 7% of patients discontinued treatment due to an adverse event; 9% of patients experienced a confirmed increase in serum creatinine of ≥0.5 mg/dl or confirmed serum phosphate of <2 mg/dl through week 48; there were no statistically significant differences between the combined tenofovir-containing arms and the entecavir arm. After 168 weeks, 16% (7/45) of the tenofovir disoproxil group, 4% (2/45) of the emtricitabine plus tenofovir disoproxil group, and 14% (3/22) of the entecavir group experienced tolerability failure. Thirteen percent (6/45) of the tenofovir disoproxil group, 13% (6/45) of the emtricitabine plus tenofovir disoproxil group, and 9% (2/22) of the entecavir group had a confirmed increase in serum creatinine ≥0.5 mg/dl or confirmed serum phosphate of <2 mg/dl.
At week 168, in this population of patients with decompensated liver disease, the rate of death was of 13% (6/45) in the tenofovir disoproxil group, 11% (5/45) in the emtricitabine plus tenofovir disoproxil group and 14% (3/22) in the entecavir group. The rate of hepatocellular carcinoma was 18% (8/45) in the tenofovir disoproxil group, 7% (3/45) in the emtricitabine plus tenofovir disoproxil group and 9% (2/22) in the entecavir group.
Subjects with a high baseline CPT score were at higher risk of developing serious adverse events (see section 4.4).
No new adverse reactions to tenofovir disoproxil were identified from a randomised, double-blind study (GS-US-174-0121) in which 280 lamivudine-resistant patients received treatment with tenofovir disoproxil (n=141) or emtricitabine/tenofovir disoproxil (n=139) for 240 weeks.
The adverse reactions with suspected (at least possible) relationship to treatment are listed below by body system organ class and frequency. Within each frequency grouping, undesirable effects are presented in order of decreasing seriousness. Frequencies are defined as very common (≥1/10), common (≥1/100 to <1/10), uncommon (≥1/1,000 to <1/100) or rare (≥1/10,000 to <1/1,000).
Table 2. Tabulated summary of adverse reactions associated with tenofovir disoproxil based on clinical study and post-marketing experience:
Very common: hypophosphataemia1
Uncommon: hypokalaemia1
Rare: lactic acidosis
Very common: dizziness
Common: headache
Very common: diarrhoea, vomiting, nausea
Common: abdominal pain, abdominal distension, flatulence
Uncommon: pancreatitis
Common: increased transaminases
Rare: hepatic steatosis, hepatitis
Very common: rash
Rare: angioedema
Uncommon: rhabdomyolysis1, muscular weakness1
Rare: osteomalacia (manifested as bone pain and infrequently contributing to fractures)1,2, myopathy1
Uncommon: increased creatinine, proximal renal tubulopathy (including Fanconi syndrome)
Rare: acute renal failure, renal failure, acute tubular necrosis, nephritis (including acute interstitial nephritis)2, nephrogenic diabetes insipidus
Very common: asthenia
Common: fatigue
1 This adverse reaction may occur as a consequence of proximal renal tubulopathy. It is not considered to be causally associated with tenofovir disoproxil in the absence of this condition.
2 This adverse reaction was identified through post-marketing surveillance but not observed in randomised controlled clinical trials or the tenofovir disoproxil expanded access program. The frequency category was estimated from a statistical calculation based on the total number of patients exposed to tenofovir disoproxil in randomised controlled clinical trials and the expanded access program (n=7,319).
As tenofovir disoproxil may cause renal damage monitoring of renal function is recommended (see sections 4.4 and 4.8 Summary of the safety profile). Proximal renal tubulopathy generally resolved or improved after tenofovir disoproxil discontinuation. However, in some patients, declines in creatinine clearance did not completely resolve despite tenofovir disoproxil discontinuation. Patients at risk of renal impairment (such as patients with baseline renal risk factors, advanced HIV disease, or patients receiving concomitant nephrotoxic medications) are at increased risk of experiencing incomplete recovery of renal function despite tenofovir disoproxil discontinuation (see section 4.4).
Co-administration of tenofovir disoproxil and didanosine is not recommended as it results in a 40-60% increase in systemic exposure to didanosine that may increase the risk of didanosine-related adverse reactions (see section 4.5). Rarely, pancreatitis and lactic acidosis, sometimes fatal, have been reported.
Weight and levels of blood lipids and glucose may increase during antiretroviral therapy (see section 4.4).
In HIV infected patients with severe immune deficiency at the time of initiation of CART, an inflammatory reaction to asymptomatic or residual opportunistic infections may arise. Autoimmune disorders (such as Graves' disease and autoimmune hepatitis) have also been reported; however, the reported time to onset is more variable and these events can occur many months after initiation of treatment (see section 4.4).
Cases of osteonecrosis have been reported, particularly in patients with generally acknowledged risk factors, advanced HIV disease or long-term exposure to CART. The frequency of this is unknown (see section 4.4).
Exacerbations of hepatitis during treatment: In studies with nucleoside-naïve patients, on-treatment ALT elevations >10 times ULN (upper limit of normal) and >2 times baseline occurred in 2.6% of tenofovir disoproxil-treated patients. ALT elevations had a median time to onset of 8 weeks, resolved with continued treatment, and, in a majority of cases, were associated with a ≥2 log10 copies/ml reduction in viral load that preceded or coincided with the ALT elevation. Periodic monitoring of hepatic function is recommended during treatment (see section 4.4).
Exacerbations of hepatitis after discontinuation of treatment: In HBV infected patients, clinical and laboratory evidence of exacerbations of hepatitis have occurred after discontinuation of HBV therapy (see section 4.4).
Assessment of adverse reactions is based on two randomised trials (studies GS-US-104-0321 and GS-US-104-0352) in 184 HIV-1 infected paediatric patients (aged 2 to <18 years) who received treatment with tenofovir disoproxil (n=93) or placebo/active comparator (n=91) in combination with other antiretroviral agents for 48 weeks (see section 5.1). The adverse reactions observed in paediatric patients who received treatment with tenofovir disoproxil were consistent with those observed in clinical studies of tenofovir disoproxil in adults (see section 4.8 Tabulated summary of adverse reactions and 5.1).
Reductions in BMD have been reported in paediatric patients. In HIV-1 infected adolescents, the BMD Z-scores observed in subjects who received tenofovir disoproxil were lower than those observed in subjects who received placebo. In HIV-1 infected children, the BMD Z-scores observed in subjects who switched to tenofovir disoproxil were lower than those observed in subjects who remained on their stavudine- or zidovudine-containing regimen (see sections 4.4 and 5.1).
In study GS-US-104-0352, 8 out of 89 paediatric patients (9.0%) exposed to tenofovir disoproxil (median tenofovir disoproxil exposure 331 weeks) discontinued study drug due to renal adverse events. Five subjects (5.6%) had laboratory findings clinically consistent with proximal renal tubulopathy, 4 of whom discontinued tenofovir disoproxil therapy. Seven patients had estimated glomerular filtration rate (GFR) values between 70 and 90 mL/min/1.73 m². Among them, 3 patients experienced a clinically meaningful decline in estimated GFR which improved after discontinuation of tenofovir disoproxil.
Assessment of adverse reactions is based on one randomised study (study GS-US-174-0115) in 106 adolescent patients (12 to <18 years of age) with chronic hepatitis B receiving treatment with tenofovir disoproxil 245 mg (n=52) or placebo (n=54) for 72 weeks. The adverse reactions observed in adolescent patients who received treatment with tenofovir disoproxil were consistent with those observed in clinical studies of tenofovir disoproxil in adults (see section 4.8 Tabulated summary of adverse reactions and 5.1).
Reductions in BMD have been observed in HBV infected adolescents. The BMD Z-scores observed in subjects who received tenofovir disoproxil were lower than those observed in subjects who received placebo (see sections 4.4 and 5.1).
Tenofovir disoproxil has not been studied in patients over the age of 65. Elderly patients are more likely to have decreased renal function, therefore caution should be exercised when treating elderly patients with tenofovir disoproxil (see section 4.4).
Since tenofovir disoproxil can cause renal toxicity, close monitoring of renal function is recommended in adult patients with renal impairment treated with Tenofovir disoproxil Mylan (see sections 4.2, 4.4 and 5.2). The use of tenofovir disoproxil is not recommended in paediatric patients with renal impairment (see sections 4.2 and 4.4).
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed in Appendix V.
Not applicable.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.