TORISEL Concentrate and solvent for solution for infusion Ref.[9095] Active ingredients: Temsirolimus
Source: European Medicines Agency (EU) Revision Year: 2019 Publisher: Pfizer Europe MA EEIG, Boulevard de la Plaine 17, 1050, Bruxelles, Belgium
Contraindications
Hypersensitivity to temsirolimus, its metabolites (including sirolimus), polysorbate 80, or to any of the excipients listed in section 6.1.
Use of temsirolimus in patients with MCL with moderate or severe hepatic impairment.
Special warnings and precautions for use
The incidence and severity of adverse events is dose-dependent. Patients receiving the starting dose of 175 mg weekly for the treatment of MCL must be followed closely to decide on dose reductions/delays.
Paediatric population
Temsirolimus is not recommended for use in paediatric patients (see sections 4.2, 4.8 and 5.1).
Elderly
Based on the results of a Phase 3 study in RCC, elderly patients (≥65 years of age) may be more likely to experience certain adverse reactions, including oedema, diarrhoea, and pneumonia. Based on the results of a Phase 3 study in MCL, elderly patients (≥65 years of age) may be more likely to experience certain adverse reactions, including pleural effusion, anxiety, depression, insomnia, dyspnoea, leukopenia, lymphopenia, myalgia, arthralgia, taste loss, dizziness, upper respiratory infection, mucositis, and rhinitis.
Renal impairment/renal failure
Temsirolimus elimination by the kidneys is negligible; studies in patients with varying renal impairment have not been conducted (see sections 4.2 and 5.2). Temsirolimus has not been studied in patients undergoing haemodialysis.
Renal failure (including fatal outcomes) has been observed in patients receiving temsirolimus for advanced RCC and/or with pre-existing renal insufficiency (see section 4.8).
Hepatic impairment
Caution should be used when treating patients with hepatic impairment.
Temsirolimus is cleared predominantly by the liver. In an open-label, dose-escalation Phase 1 study in 110 subjects with advanced malignancies and either normal or impaired hepatic function, concentrations of temsirolimus and its metabolite sirolimus were increased in patients with elevated aspartate aminotransferase (AST) or bilirubin levels. Assessment of AST and bilirubin levels is recommended before initiation of temsirolimus and periodically after. An increased rate of fatal events was observed in patients with moderate and severe hepatic impairment. The fatal events included those due to progression of disease; however a causal relationship cannot be excluded.
Based on the Phase 1 study, no dose adjustment of temsirolimus is recommended for RCC patients with baseline platelet counts ≥100 x 109/l and mild to moderate hepatic impairment (total bilirubin up to 3 times upper limit of normal [ULN] with any abnormality of AST, or as defined by Child-Pugh Class A or B). For patients with RCC and severe hepatic impairment (total bilirubin >3 times ULN with any abnormality of AST, or as defined by Child-Pugh Class C), the recommended dose for patients who have baseline platelets ≥100 x 109/l is 10 mg intravenous once a week infused over a 30-60 minute period (see section 4.2).
Intracerebral bleeding
Patients with central nervous system (CNS) tumours (primary CNS tumours or metastases) and/or receiving anticoagulation therapy may be at an increased risk of developing intracerebral bleeding (including fatal outcomes) while receiving therapy with temsirolimus.
Thrombocytopenia, neutropenia, and anaemia
Grades 3 and 4 thrombocytopenia and/or neutropenia have been observed in the MCL clinical trial (see section 4.8). Patients on temsirolimus who develop thrombocytopenia may be at increased risk of bleeding events, including epistaxis (see section 4.8). Patients on temsirolimus with baseline neutropenia may be at risk of developing febrile neutropenia. Cases of anaemia have been reported in RCC and MCL (see section 4.8). Monitoring of complete blood count is recommended prior to initiating temsirolimus therapy and peridically thereafter.
Infections
Patients may be immunosuppressed and should be carefully observed for the occurrence of infections, including opportunistic infections. Among patients receiving 175 mg/week for the treatment of MCL, infections (including Grade 3 and 4 infections) were substantially increased compared to lower doses and compared to conventional chemotherapy. Cases of pneumocystis jiroveci pneumonia (PCP) some with fatal outcomes, have been reported in patients who received temsirolimus, many of whom also received corticosteroids or other immunosuppressive agents. Prophylaxis of PCP should be considered for patients who require concomitant use of corticosteroids or other immunosuppressive agents based upon current standard of care.
Cataracts
Cataracts have been observed in some patients who received the combination of temsirolimus and interferon-α (IFN-α).
Hypersensitivity/infusion reactions
Hypersensitivity/infusion reactions (including some life-threatening and rare fatal reactions), including and not limited to flushing, chest pain, dyspnoea, hypotension, apnoea, loss of consciousness, hypersensitivity and anaphylaxis, have been associated with the administration of temsirolimus (see section 4.8). These reactions can occur very early in the first infusion, but may also occur with subsequent infusions. Patients should be monitored early during the infusion and appropriate supportive care should be available. The temsirolimus infusion should be interrupted in all patients with severe infusion reactions and appropriate medical therapy administered. A benefit-risk assessment should be done prior to the continuation of temsirolimus therapy in patients with severe or life-threatening reactions.
If a patient develops a hypersensitivity reaction during the temsirolimus infusion despite the premedication, the infusion must be stopped and the patient observed for at least 30 to 60 minutes (depending on the severity of the reaction). At the discretion of the physician, treatment may be resumed after the administration of an H1-receptor antagonist (diphenhydramine or similar antihistamine) and a H2-receptor antagonist (intravenous famotidine 20 mg or intravenous ranitidine 50 mg) approximately 30 minutes before restarting the temsirolimus infusion. Administration of corticosteroids may be considered; however, the efficacy of corticosteroid treatment in this setting has not been established. The infusion may then be resumed at a slower rate (up to 60 minutes) and should be completed within six hours from the time that temsirolimus is first added to sodium chloride 9 mg/ml (0.9%) solution for injection.
Because it is recommended that an H1 antihistamine be administered to patients before the start of the intravenous temsirolimus infusion, temsirolimus should be used with caution in patients with known hypersensitivity to the antihistamine or in patients who cannot receive the antihistamine for other medical reasons.
Hypersensitivity reactions, including anaphylactic/anaphylactoid reactions, angioedema, exfoliative dermatitis and hypersensitivity vasculitis, have been associated with the oral administration of sirolimus.
Hyperglycaemia/glucose intolerance/diabetes mellitus
Patients should be advised that treatment with temsirolimus may be associated with an increase in blood glucose levels in diabetic and non-diabetic patients. In the RCC clinical trial, a Phase 3 clinical trial for RCC, 26% of patients reported hyperglycaemia as an adverse event. In the MCL clinical trial, a Phase 3 clinical trial for MCL, 11% of patients reported hyperglycaemia as an adverse event. This may result in the need for an increase in the dose of, or initiation of, insulin and/or hypoglycaemic agent therapy. Patients should be advised to report excessive thirst or any increase in the volume or frequency of urination.
Interstitial lung disease
There have been cases of non-specific interstitial pneumonitis, including fatal reports, occurring in patients who received weekly intravenous temsirolimus. Some patients were asymptomatic or had minimal symptoms with pneumonitis detected on computed tomography scan or chest radiograph. Others presented with symptoms such as dyspnoea, cough, and fever. Some patients required discontinuation of temsirolimus or treatment with corticosteroids and/or antibiotics, while some patients continued treatment without additional intervention. It is recommended that patients undergo baseline radiographic assessment by lung computed tomography scan or chest radiograph prior to the initiation of temsirolimus therapy. Periodical follow-up assessments may be considered. It is recommended that patients be followed closely for occurrence of clinical respiratory symptoms and patients should be advised to report promptly any new or worsening respiratory symptoms. If clinically significant respiratory symptoms develop, temsirolimus administration may be withheld until after recovery of symptoms and improvement of radiographic findings related to pneumonitis. Opportunistic infections such as PCP should be considered in the differential diagnosis. Empiric treatment with corticosteroids and/or antibiotics may be considered. For patients who require use of corticosteroids, prophylaxis of PCP should be considered based upon current standard of care.
Hyperlipaemia
The use of temsirolimus was associated with increases in serum triglycerides and cholesterol. In the RCC clinical trial 1, hyperlipaemia was reported as an adverse event in 27% of patients. In the MCL clinical trial, hyperlipaemia was reported as an adverse event in 9.3% of patients. This may require initiation, or increase, in the dose of lipid-lowering agents. Serum cholesterol and triglycerides should be tested before and during treatment with temsirolimus. The known association of temsirolimus with hyperlipaemia may predispose to myocardial infarction.
Wound healing complications
The use of temsirolimus has been associated with abnormal wound healing; therefore, caution should be exercised with the use of temsirolimus in the peri-surgical period.
Malignancies
The possible development of lymphoma and other malignancies, particularly of the skin, may result from immunosuppression. As usual for patients with increased risk for skin cancer, exposure to sunlight and ultraviolet (UV) light should be limited by wearing protective clothing and using a sunscreen with a high protection factor.
Concomitant use of temsirolimus with sunitinib
The combination of temsirolimus and sunitinib resulted in dose-limiting toxicity. Dose-limiting toxicities (Grade ¾ erythematous maculopapular rash, gout/cellulitis requiring hospitalisation) were observed in 2 out of 3 patients treated in the first cohort of a Phase 1 study at doses of temsirolimus 15 mg intravenous per week and sunitinib 25 mg oral per day (Days 1-28 followed by a 2-week rest) (see section 4.5).
Concomitant use of angiotensin-converting enzyme (ACE) inhibitors and/or calcium channel blockers
Caution should be exercised when temsirolimus is given concomitantly with ACE inhibitors (e.g. ramipril) and/or calcium channel blockers (e.g. amlodipine). An increased risk of angioneurotic oedema (including delayed reactions occurring two months following initiation of therapy) is possible in patients who receive temsirolimus concomitantly with an ACE inhibitor and/or a calcium channel blocker (see sections 4.5 and 4.8).
Agents inducing CYP3A metabolism
Agents such as carbamazepine, phenobarbital, phenytoin, rifampicin, and St. John's wort are strong inducers of CYP3A4/5 and may decrease composite exposure of the active drug substances, temsirolimus and its metabolite, sirolimus. Therefore, for patients with RCC, continuous administration beyond 5-7 days with agents that have CYP3A4/5 induction potential should be avoided. For patients with MCL, it is recommended that coadministration of CYP3A4/5 inducers should be avoided due to the higher dose of temsirolimus (see section 4.5).
Agents inhibiting CYP3A metabolism
Agents such as protease inhibitors (nelfinavir, ritonavir), antifungals (e.g. itraconazole, ketoconazole, voriconazole), and nefazodone are strong CYP3A4 inhibitors and may increase blood concentrations of the active drug substances, temsirolimus and its metabolite, sirolimus. Therefore, concomitant treatment with agents that have strong CYP3A4 inhibition potential should be avoided. Concomitant treatment with moderate CYP3A4 inhibitors (e.g. aprepitant, erythromycin, fluconazole, verapamil, grapefruit juice) should only be administered with caution in patients receiving 25 mg and should be avoided in patients receiving temsirolimus doses higher than 25 mg (see section 4.5). Alternative treatments with agents that do not have CYP3A4 inhibition potential should be considered (see section 4.5).
Vaccinations
Immunosuppressants may affect responses to vaccination. During treatment with temsirolimus, vaccination may be less effective. The use of live vaccines should be avoided during treatment with temsirolimus. Examples of live vaccines are: measles, mumps, rubella, oral polio, Bacillus Calmette- Guérin (BCG), yellow fever, varicella, and TY21a typhoid vaccines.
Excipients
After first dilution of the concentrate with 1.8 ml of the supplied solvent, the concentrate-solvent mixture contains 35% volume ethanol (alcohol), i.e., up to 0.693 g per 25 mg dose of temsirolimus, equivalent to 17.6 ml beer or 7.3 ml wine per dose. Patients administered the higher dose of 175 mg of temsirolimus for the initial treatment of MCL may receive up to 4.85 g of ethanol (equivalent to 123 ml beer or 51 ml wine per dose).
Harmful for those suffering from alcoholism.
To be taken into account in pregnant or breast-feeding women, children and high-risk groups, such as patients with liver disease or epilepsy. The amount of alcohol in this medicinal product may alter the effects of other medicines. The amount of alcohol in this medicinal product may impair the ability to drive or use machines (see section 4.7).
Interaction with other medicinal products and other forms of interaction
Interaction studies have only been performed in adults.
Concomitant use of temsirolimus with sunitinib
The combination of temsirolimus and sunitinib resulted in dose-limiting toxicity. Dose-limiting toxicities (Grade ¾ erythematous maculopapular rash, gout/cellulitis requiring hospitalisation) were observed in 2 out of 3 patients treated in the first cohort of a Phase 1 study at doses of temsirolimus 15 mg intravenous per week and sunitinib 25 mg oral per day (Days 1-28 followed by a 2-week rest) (see section 4.4).
Concomitant use of angiotensin-converting enzyme (ACE) inhibitors and/or calcium channel blockers
An increased incidence of angioneurotic oedema (including delayed reactions occurring two months following initiation of therapy) has been observed in patients who received temsirolimus or other mTOR inhibitors in combination with an ACE inhibitor (e.g. ramipril) and/or a calcium channel blocker (e.g. amlodipine) (see sections 4.4 and 4.8).
Agents inducing CYP3A metabolism
Co-administration of temsirolimus with rifampicin, a potent CYP3A4/5 inducer, had no significant effect on temsirolimus maximum concentration (Cmax) and area under the concentration vs. time curve (AUC) after intravenous administration, but decreased sirolimus Cmax by 65% and AUC by 56%, compared to temsirolimus treatment alone. Therefore, concomitant treatment with agents that have CYP3A4/5 induction potential should be avoided (e.g. carbamazepine, phenobarbital, phenytoin, rifampicin, and St. John's wort) (see section 4.4).
Agents inhibiting CYP3A metabolism
Co-administration of temsirolimus 5 mg with ketoconazole, a potent CYP3A4 inhibitor, had no significant effect on temsirolimus C max or AUC; however, sirolimus AUC increased 3.1-fold, and AUC sum (temsirolimus + sirolimus) increased 2.3-fold compared to temsirolimus alone. The effect on the unbound concentrations of sirolimus has not been determined, but is expected to be larger than the effect on whole-blood concentrations due to the saturable binding to red blood cells. The effect may also be more pronounced at a 25 mg dose. Therefore, substances that are potent inhibitors of CYP3A4 activity (e.g. nelfinavir, ritonavir, itraconazole, ketoconazole, voriconazole, nefazodone) increase sirolimus blood concentrations. Concomitant treatment of temsirolimus with these agents should be avoided (see section 4.4).
Concomitant treatment with moderate CYP3A4 inhibitors (e.g., diltiazem, verapamil, clarithromycin, erythromycin, aprepitant, amiodarone) should only be administered with caution in patients receiving 25 mg and should be avoided in patients receiving temsirolimus doses higher than 25 mg.
Interaction with medicinal products metabolised by CYP2D6 or CYP3A4/5
In 23 healthy subjects the concentration of desipramine, a CYP2D6 substrate, was unaffected when 25 mg of temsirolimus was co-administered. In 36 patients with MCL, including 4 poor metabolisers, the effect of CYP2D6 inhibition after administration of single doses of 175 mg and 75 mg temsirolimus was investigated. Population PK analysis based on sparse sampling indicated no clinically significant interaction effect on AUC and Cmax of the CYP2D6 substrate desipramine. No clinically significant effect is anticipated when temsirolimus is co-administered with agents that are metabolised by CYP2D6.
The effect of a 175 or 75 mg temsirolimus dose on CYP3A4/5 substrates has not been studied. However, in vitro studies in human liver microsomes followed by physiologically-based pharmacokinetic modelling indicate that the blood concentrations achieved after a 175 mg dose of temsirolimus most likely leads to relevant inhibition of CYP3A4/5 (see section 5.2). Therefore, caution is advised during concomitant administration of temsirolimus at a dose of 175 mg with medicinal products that are metabolised predominantly via CYP3A4/5 and that have a narrow therapeutic index.
Interactions with medicinal products that are P-glycoprotein substrates
In an in vitro study, temsirolimus inhibited the transport of P-glycoprotein (P-gp) substrates with an IC50 value of 2 μM. In vivo, the effect of P-gp inhibition has not been investigated in a clinical drug-drug interaction study, however, recent preliminary data from a Phase 1 study of combined lenalidomide (dose of 25 mg) and temsirolimus (dose of 20 mg) seem to support the in vitro findings and suggest an increased risk of adverse events. Therefore, when temsirolimus is co-administered with medicinal products which are P-gp substrates (e.g. digoxin, vincristine, colchicine, dabigatran, lenalidomide, and paclitaxel) close monitoring for adverse events related to the co-administered medicinal products should be observed.
Amphiphilic agents
Temsirolimus has been associated with phospholipidosis in rats. Phospholipidosis has not been observed in mice or monkeys treated with temsirolimus, nor has it been documented in patients treated with temsirolimus. Although phospholipidosis has not been shown to be a risk for patients administered temsirolimus, it is possible that combined administration of temsirolimus with other amphiphilic agents such as amiodarone or statins could result in an increased risk of amphiphilic pulmonary toxicity.
Fertility, pregnancy and lactation
Women of childbearing potential/Contraception in males and females
Due to the unknown risk related to potential exposure during early pregnancy, women of childbearing potential must be advised not to become pregnant while using Torisel.
Men with partners of childbearing potential should use medically acceptable contraception while receiving Torisel (see section 5.3).
Pregnancy
There are no adequate data from the use of temsirolimus in pregnant women. Studies in animals have shown reproductive toxicity. In reproduction studies in animals, temsirolimus caused embryo/foetotoxicity that was manifested as mortality and reduced foetal weights (with associated delays in skeletal ossification) in rats and rabbits. Teratogenic effects (omphalocele) were seen in rabbits (see section 5.3).
The potential risk for humans is unknown. Torisel must not be used during pregnancy, unless the risk for the embryo is justified by the expected benefit for the mother.
Breast-feeding
It is unknown whether temsirolimus is excreted in human breast milk. The excretion of temsirolimus in milk has not been studied in animals. However, sirolimus, the main metabolite of temsirolimus, is excreted in milk of lactating rats. Because of the potential for adverse reactions in breast-fed infants from temsirolimus, breast-feeding should be discontinued during therapy.
Fertility
In male rats, decreased fertility and partly reversible reductions in sperm counts were reported (see section 5.3).
Effects on ability to drive and use machines
Temsirolimus has no or negligible influence on the ability to drive and use machines based on the evidence available.
For patients receiving the higher dose of 175 mg intravenous of temsirolimus for the treatment of MCL, the amount of ethanol in this medicinal product may impair the ability to drive or use machines (see section 4.4).
Undesirable effects
Summary of the safety profile
The most serious reactions observed with temsirolimus in clinical trials are hypersensitivity/infusion reactions (including some life-threatening and rare fatal reactions), hyperglycaemia/glucose intolerance, infections, interstitial lung disease (pneumonitis), hyperlipaemia, intracranial haemorrhage, renal failure, intestinal perforation, wound healing complication, thrombocytopenia, neutropenia (including febrile neutropenia), pulmonary embolism.
The adverse reactions (all grades) experienced by at least 20% of the patients in RCC and MCL registration studies include anaemia, nausea, rash (including rash, pruritic rash, maculopapular rash, pustular rash), decreased appetite, oedema asthenia, fatigue, thrombocytopenia, diarrhoea, pyrexia, epistaxis, mucosal inflammation, stomatitis, vomiting, hyperglycaemia, hypercholesterolemia, dysgeusia, pruritus, cough, infection, pneumonia, dyspnoea.
Cataracts have been observed in some patients who received the combination of temsirolimus and IFN-α.
Based on the results of the phase 3 studies, elderly patients may be more likely to experience certain adverse reactions, including face oedema, pneumonia, pleural effusion, anxiety, depression, insomnia, dyspnoea, leukopenia, lymphopenia, myalgia, arthralgia, ageusia, dizziness, upper respiratory infection, mucositis, and rhinitis.
Serious adverse reactions observed in clinical trials of temsirolimus for advanced RCC, but not in clinical trials of temsirolimus for MCL include: anaphylaxis, impaired wound healing, renal failure with fatal outcomes, and pulmonary embolism.
Serious adverse reactions observed in clinical trials of temsirolimus for MCL, but not in clinical trials of temsirolimus for advanced RCC include: thrombocytopenia, and neutropenia (including febrile neutropenia).
See section 4.4 for additional information concerning serious adverse reactions, including appropriate actions to be taken if specific reactions occur.
The occurrence of undesirable effects following the dose of 175 mg temsirolimus/week for MCL, e.g. Grade 3 or 4 infections or thrombocytopenia, is associated with a higher incidence than that observed with either 75 mg temsirolimus/week or conventional chemotherapy.
Tabulated list of adverse reactions
Adverse reactions that were reported in RCC and MCL patients in the phase 3 studies are listed below (Table 1), by system organ class, frequency and grade of severity (NCI-CTCAE). Frequencies are defined as follows: very common (≥1/10), common (≥1/100 to <1/10), uncommon (≥1/1,000 to <1/100), rare (1/10,000 to <1/1,000), very rare (<1/10,000) and not known (cannot be estimated from the available data).Within each frequency grouping, adverse reactions are presented in order of decreasing seriousness.
Table 1. Adverse reactions from clinical trials in RCC (study 3066K1-304) and in MCL (study 3066K1-305):
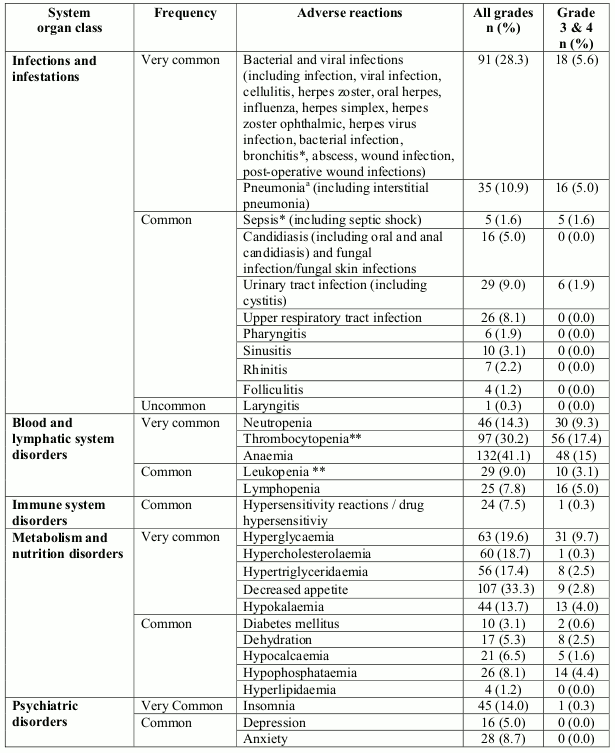
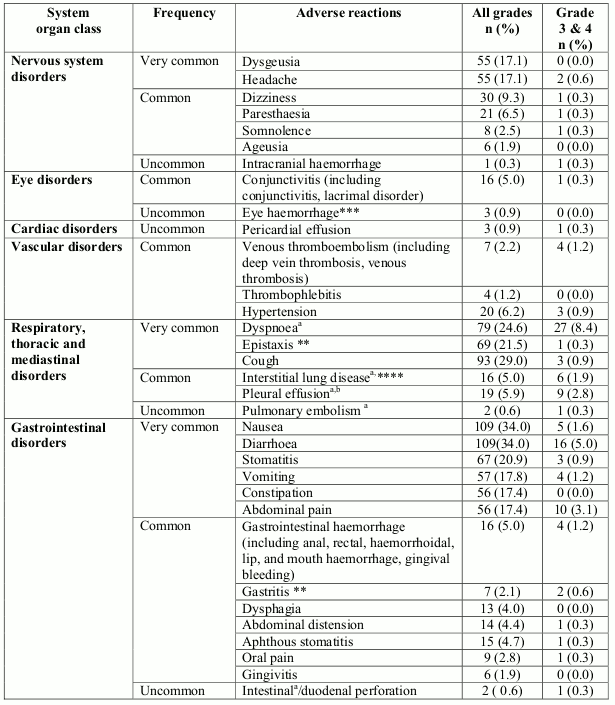
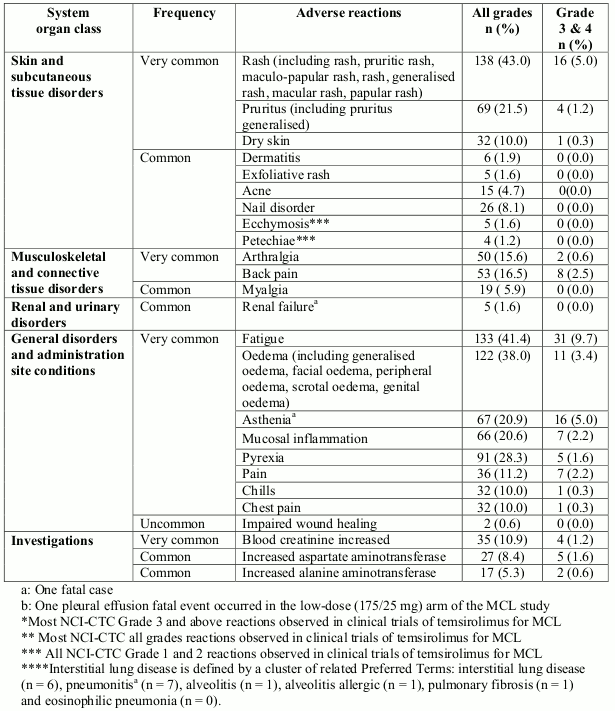
Adverse reactions that were reported in post-marketing experience are listed below (Table 2).
Table 2. Adverse reactions reported in post-marketing setting:
Infections and infestations
Rare: Pneumocystis jiroveci pneumonia
Immune system disorders
Not known: Angioneurotic oedema-type reactions
Skin and subcutaneous tissue disorders
Not known: Stevens-Johnson syndrome
Musculoskeletal and connective tissue disorders
Not known: Rhabdomyolysis
Description of selected adverse reactions
Post-marketing experience
Angioneurotic oedema-type reactions have been reported in some patients who received temsirolimus and ACE-inhibitors concomitantly.
Cases of PCP, some with fatal outcomes, have been reported (see section 4.4).
Paediatric population
In a Phase 1/2 study, 71 patients (59 patients, aged from 1 to 17 years old, and 12 patients, aged 18 to 21 years) were administered temsirolimus at doses ranging from 10 mg/m² to 150 mg/m² (see section 5.1).
The adverse reactions reported by the highest percentage of patients were haematologic (anaemia, leukopenia, neutropenia, and thrombocytopenia), metabolic (hypercholesterolemia, hyperlipaemia, hyperglycaemia, increase of serum aspartate amino transferase (AST) and serum alanine aminotransferase (ALT) plasma levels), and digestive (mucositis, stomatitis, nausea, and vomiting).
Reporting of suspected adverse reactions
Reporting suspected adverse reactions after authorisation of the medicinal product is important. It allows continued monitoring of the benefit/risk balance of the medicinal product. Healthcare professionals are asked to report any suspected adverse reactions via the national reporting system listed in Appendix V.
Incompatibilities
This medicinal product must not be mixed with other medicinal products, except those mentioned in section 6.6.
Torisel 30 mg concentrate must not be added directly to aqueous infusion solutions. Direct addition of Torisel 30 mg concentrate to aqueous solutions will result in precipitation of medicinal product.
Always dilute Torisel 30 mg concentrate with 1.8 ml of the supplied solvent before adding to the infusion solution. The concentrate-solvent mixture may only be administered in sodium chloride 9 mg/ml (0.9%) solution for injection.
Torisel, when diluted, contains polysorbate 80, which is known to increase the rate of di-(2-ethylhexyl) phthalate extraction (DEHP) from polyvinyl chloride (PVC). This incompatibility has to be considered during the preparation and administration of Torisel. It is important that the recommendations in sections 4.2 and 6.6 be followed closely.
PVC bags and medical devices must not be used for the administration of preparations containing polysorbate 80, because polysorbate 80 leaches DEHP from PVC.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.