TOVEDESO Prolonged-release tablet Ref.[7701] Active ingredients: Desfesoterodine
Source: Medicines & Healthcare Products Regulatory Agency (GB) Revision Year: 2017 Publisher: TEVA UK Limited, Brampton Road, Hampden Park, Eastbourne, East Sussex, BN22 9AG, United Kingdom
Pharmacodynamic properties
Pharmacotherapeutic group: Urologicals, Urinary antispasmodics
ATC code: G04BD13
Mechanism of action
Desfesoterodine is a competitive, specific muscarinic receptor antagonist. It is the primary active metabolite of fesoterodine, and regarded as main active pharmacological principle of fesoterodine; fesoterodine is considered as prodrug of desfesoterodine.
Fesoterodine fumarate is rapidly and extensively hydrolyzed by non-specific esterases to desfesoterodine, 5-hydroxymethyl derivative (5-HMT). The mechanism of action of fesoterodine is a blockade of muscarinic receptors (M1-M5). The affinity of desfesoterodine for the muscarinic receptors is 2 orders of magnitude greater than that of fesoterodine. Since desfesoterodine is rapidly formed from fesoterodine by ester cleavage, in most species, fesoterodine cannot be detected in blood plasma; however, desfesoterodine can be detected. This is the case in several species including human and mouse, the mouse being the most relevant nonclinical species which most resembles the PK situation in humans. In both species, fesoterodine functionally acts as a pro-drug and toxicity is triggered by antimuscarinic effects.
Clinical efficacy and safety
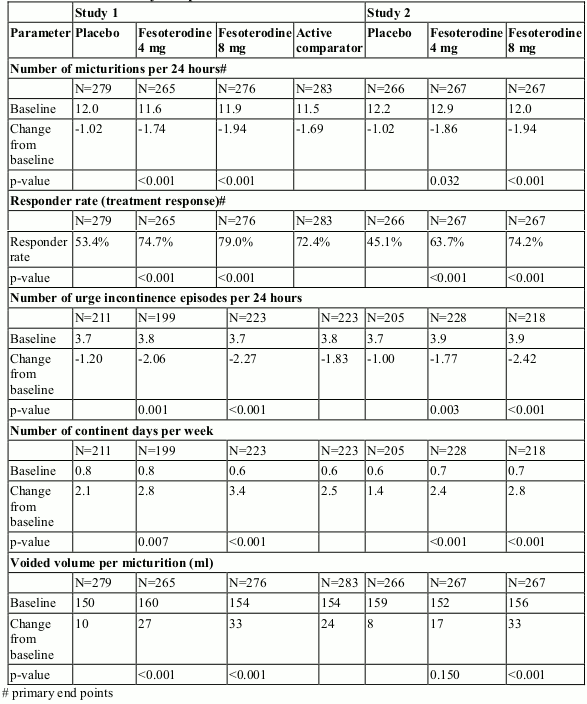
Since fesoterodine derives its pharmacological activity from its metabolite, desfesoterodine, fesoterodine data are directly relevant to the efficacy and safety of desfesoterodine. The efficacy of fixed doses of fesoterodine 4 mg and 8 mg was evaluated in two Phase 3 randomised, double-blind, placebocontrolled, 12-week studies. Female (79%) and male (21%) patients with a mean age of 58 years (range 19-91 years) were included. A total of 33% of patients were ≥65 years of age and 11% were ≥75 years of age.
Fesoterodine treated patients had statistically significant mean reductions in the number of micturitions per 24 hours and in the number of urge incontinence episodes per 24 hours at the end of treatment compared to placebo. Likewise, the response rate (% of patients reporting that their condition has been "greatly improved" or "improved" using a 4-point Treatment Benefit Scale) was significantly greater with fesoterodine compared to placebo. Furthermore, fesoterodine improved the mean change in the voided volume per micturition, and the mean change in the number of continent days per week (see Table 1 below).
Table 1. Mean changes from Baseline to end of treatment for primary and selected secondary endpoints:
Cardiac electrophysiology
The effect of fesoterodine, prodrug of desfesoterodine, 4 mg and 28 mg on the QT interval was thoroughly evaluated in a double-blind, randomised, placeboand positive-controlled (moxifloxacin 400 mg) parallel group study with oncedaily treatment over a period of 3 days in 261 male and female subjects aged 45 to 65 years. Change from baseline in QTc based on the Fridericia correction method did not show any differences between the active treatment and placebo group.
Pharmacokinetic properties
Desfesoterodine is the active metabolite of the pro-drug fesoterodine.
Absorption
After oral administration of fesoterodine, due to rapid and extensive hydrolysis by non-specific plasma esterases, the parent compound was not measurable in plasma.
Pharmacokinetics of Desfesoterodine have shown to be equivalent after administration of oral fesoterodine and desfesoterodine in equimolar doses under fasted and fed conditions.
Due to the first pass metabolism, bioavailability of the active metabolite desfesoterodine is 52% following oral administration of fesoterodine.
Pharmacokinetics of desfesoterodine are linear after single dose oral administration in doses of 3.5 and 7 mg. Maximum plasma levels of desfesoterodine are reached approximately 5 hours after administration of a desfesoterodine prolonged release formulation. Therapeutic plasma levels are achieved after the first administration of desfesoterodine. No accumulation occurs after multiple-dose administration.
Exposure to desfesoterodine was increased about 5-10% in the fed state as compared to the fasted state following oral administration of desfesoterodine.
Although bioequivalence between the 2 treatments could not be demonstrated the difference is considered not to be clinically relevant. Therefore, dose adjustment with regard to food is not necessary with respect to fesoterodine.
Fesoterodine and desfesoterodine show comparable pharmacokinetics following drug administration in fed state and under fasting conditions, respectively.
Distribution
Plasma protein binding of desfesoterodine is low with approximately 50% bound to albumin and alpha-1-acid glycoprotein. The mean steady-state volume of distribution following intravenous infusion of desfesoterodine is 169 l.
Biotransformation
After oral administration of fesoterodine, the parent compound is rapidly and extensively hydrolysed to its active metabolite desfesoterodine.
Desfesoterodine is further metabolised in the liver to its carboxy metabolite and N-desisopropyl metabolite with involvement of CYP2D6 and CYP3A4 respectively. Furthermore, CYP2A6 may be involved in the formation of the N-desisopropyl metabolite. Mean Cmax and AUC of the active metabolite are 1.7 and 2-fold higher, respectively, in CYP2D6 poor metabolisers as compared to extensive metabolisers.
No in vivo interconversion of the R-enantiomer was observed after the administration of R-desfesoterodine in a prolonged release formulation.
Elimination
Hepatic metabolism and renal excretion contribute significantly to the elimination of the active metabolite. After oral administration of fesoterodine, approximately 70% of the administered dose was recovered in urine as the active metabolite (16%), carboxy metabolite (34%), carboxy-N-desisopropyl metabolite (18%), or N-desisopropyl metabolite (1%), and a smaller amount (7%) was recovered in faeces. The terminal half-life of desfesoterodine following oral administration of fesoterodine is approximately 7 hours and is absorption rate-limited.
Age and gender
No dose adjustment is recommended in these subpopulations. The pharmacokinetics of desfesoterodine following oral administration of fesoterodine are not significantly influenced by age and gender.
Renal impairment
In patients with mild or moderate renal impairment (GFR 30–80 ml/min), Cmax and AUC of desfesoterodine following oral administration of fesoterodine increased up to 1.5 and 1.8-fold, respectively, as compared to healthy subjects. In patients with severe renal impairment (GFR <30 ml/min), Cmax and AUC are increased 2.0 and 2.3-fold, respectively.
Hepatic impairment
In patients with moderate hepatic impairment (Child Pugh B), Cmax and AUC of desfesoterodine following oral administration of fesoterodine increased 1.4 and 2.1-fold, respectively, as compared to healthy subjects. Pharmacokinetics of fesoterodine in patients with severe hepatic impairment have not been studied.
Paediatric population
The pharmacokinetics of fesoterodine and desfesoterodine have not been evaluated in paediatric patients.
Preclinical safety data
In non-clinical safety pharmacology, general toxicity, genotoxicity and carcinogenicity studies with fesoterodine prodrug of desfesoterodine, no clinically relevant effects have been observed, except those related to the pharmacological effect of the active substance.
In 13-week oral repeat-dose toxicity study in mice, observed effects were similar for both, desfesoterodine succinate (108.88 mg/kg/day) and fesoterodine fumarate (125 mg/kg/day) molar equivalent dose groups; comparison of the toxicokinetic parameters, showed that the desfesoterodine plasma concentration-versus-time profiles were similar for these two groups. Results suggested that observed effects were due to desfesoterodine only.
Reproduction studies with fesoterodine have shown minor embryotoxicity at doses close to maternally toxic ones (increased number of resorptions, preimplantation and post-implantation losses).
Supratherapeutic concentrations of desfesoterodine, have been shown to inhibit K+ current in cloned human ether-à-go-go-related gene (hERG) channels and prolong action potential duration (70% and 90% repolarisation) in canine isolated Purkinje fibres. However in conscious dogs, it had no effect on the QT interval and QTc interval at plasma exposures at least 33-fold higher than mean peak free plasma concentration in human subjects who are extensive metabolisers and 21-fold higher than measured in subjects who are poor CYP2D6 metabolisers after fesoterodine 8 mg once daily.
In a study of fertility and early embryonic development in mice, fesoterodine had no effect on male or female fertility at the highest dose evaluated, 45 mg/kg/day. Fesoterodine had no effect on reproductive function, or early embryonic development of the fetus at non-maternally toxic doses in mice, although a slight decrease in corpora lutea, implantation sites, and viable fetuses was noted at the maternally toxic dose of 45 mg/kg/day. The maternal No-Observed-Effect Level (NOEL) and the NOEL for effects on reproduction and early embryonic development were both 15 mg/kg/day. Based on AUC, the systemic exposure was 0.6 to 1.5 times higher in mice than in humans at the MRHD, whereas based on peak plasma concentrations, the exposure in mice was 5 to 9 times higher.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.