TRYNGOLZA Solution for injection Ref.[115924] Active ingredients: Olezarsen
Source: European Medicines Agency (EU) Revision Year: 2025 Publisher: Ionis Ireland Limited, St. James House, 72 Adelaide Road, Dublin 2, D02 Y017, Ireland
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: lipid modifying agents, other lipid modifying agents
Anatomical therapeutic chemical (ATC) code: not yet assigned
Mechanism of action
Olezarsen is an antisense oligonucleotide-triantennary N-acetylgalactosamine (GalNAc 3) conjugate that causes degradation of apolipoprotein C3 (apoC-III) messenger ribonucleic acid (mRNA) through selective binding to its mRNA, which leads to ribonuclease H1 (RNase H1)-mediated cleavage of apoC-III mRNA. Olezarsen is perfectly complementary to the site on chromosome 11 positions 116, 833, 046 through 116, 833, 065, corresponding to the gene apoC-III according to Ensembl version 109 (GRCh38 build) of the homo sapiens genome. This results in specific reductions of serum apoC-III protein leading to plasma triglyceride reductions. Studies suggest that apoC-III regulates both triglyceride metabolism and hepatic clearance of chylomicrons and other triglyceride-rich lipoproteins.
Pharmacodynamic effects
Effects of olezarsen on lipid parameters
In a phase 3 clinical trial in patients with FCS (Balance trial), administration of olezarsen decreased apoC-III, triglycerides (TG), chylomicron triglycerides, apolipoprotein B-48 (apoB-48), total cholesterol (TC), and non-high-density lipoprotein cholesterol (non-HDL-C). It also increased high-density lipoprotein cholesterol (HDL), total apolipoprotein B (apoB), and low-density lipoprotein cholesterol (LDL-C). Mean LDL-C levels remained within the normal range (i.e., <70 mg/dL) for 74% of patients.
Cardio electrophysiology
At a dose 1.5-times the maximum recommended dose for olezarsen, no clinically significant corrected QT interval prolongation was observed.
Clinical efficacy and safety
The efficacy and safety of olezarsen was studied in a randomised, multicentre, double-blind, placebo-controlled clinical trial (Balance trial) that included 66 adult patients with FCS. Patients were screened and enrolled based on documented loss-of-function variants in various genes known to cause complete or partial deficiency in the function of lipoprotein lipase, an enzyme that hydrolyzes TGs transported by TG-rich lipoproteins into free fatty acids. After a ≥4-week run-in period where patients continued to follow a diet with ≤20 g fat per day, patients were randomly assigned 1:1 to cohort A (50 mg) or cohort B (80 mg) and each cohort was further randomised 2:1 to receive olezarsen or placebo, respectively, via subcutaneous injection over a 53-week treatment period.
The main inclusion criteria for the trial were: a diagnosis of FCS confirmed by documentation of homozygote, compound heterozygote, or double heterozygote for loss-of-function mutations in type 1-causing genes [such as Lipoprotein Lipase (LPL), Glycosylphosphatidylinositol Anchored High Density Lipoprotein Binding Protein 1 (GPIHBP1), Apolipoprotein A5 (APOA5), Apolipoprotein C2 (APOC2), Glycerol-3-Phosphate Dehydrogenase 1 (GPD1), or Lipase Maturation Factor 1 (LMF1)]; and with or without a history of pancreatitis. History of pancreatitis is defined as a recorded diagnosis of acute pancreatitis or hospitalisation for severe abdominal pain consistent with acute pancreatitis with no alternate diagnosis, within 10 years prior to screening. Enrollment of patients without a history of pancreatitis was capped at 35% (i.e. ≤21 of the 60 planned patients).
Patient demographic and baseline characteristics were generally similar across the 3 treatment groups. A total of 66 patients were enrolled. The mean age was 45 years, 38 (58%) were females, 56 (85%) were white, and 59 (89%) of non-Hispanic or Latino ethnicity. Out of a total of 66 patients, 55 (83%) had loss of function mutation in LPL gene including 40 (61%) with homozygote LPL mutation, and 11 (17%) had other causative variants in APOA5, GPIHBP1, LMF1 and APOC2 genes. The proportion of patients with diabetes at enrollment was 32% in the olezarsen 80 mg group and 14% in the olezarsen 50 mg group compared with 26% in the placebo group. Across all treatment groups, patients enrolled were being treated with statins (24%), omega-3 fatty acids (38%), fibrates (46%), or other lipid-lowering therapies (9%) at trial entry. Patients on lipid-lowering therapy had to maintain stable doses for at least 4 weeks prior to screening and remain on stable therapy throughout the trial. Additionally, all patients were to adhere to their prescribed diet for the entire duration of the trial. Seventy-one percent (71%) of all patients had a history of documented acute pancreatitis in the prior 10 years. Mean (standard deviation [SD]) fasting TG level at baseline was 2 629.5 (1 315.45) mg/dL.
Olezarsen led to a statistically significant reduction in triglyceride levels in the 80 mg group as compared to placebo at the primary efficacy endpoint, defined as percent change in fasting triglycerides from baseline to month 6 (average of weeks 23, 25, and 27), see Table 2 below. Olezarsen 50 mg is not an approved dosing regimen for FCS and further analyses are not shown.
Table 2. Mean baseline (BL) and least-squares mean percent (%) changes from baseline in lipid/lipoprotein parameters in patients with FCS at months 6 and 12 (Balance trial):
| Parameter (mg/dL) | Olezarsen 80 mg N=22 | Placebo N=23 | Olezarsen 80 mg vs. Placebo | |||||
|---|---|---|---|---|---|---|---|---|
| BL | % change month 6 | % change month 12 | BL | % change month 6 | % change month 12 | Treatment difference (95% CI) | ||
| at month 6 | at month 12 | |||||||
| Triglycerides | 2 613.1 | -32 | -39 | 2 595.7 | +12 | +21 | -43.5* (-69.1, -17.9) | -59.4† (-90.7, -28.1) |
| ApoC-III | 27.5 | -66 | -64 | 27.7 | +8 | +17 | -73.7† (-94.6, -52.8) | -81.3† (-104.7, -57.9) |
| ApoB-48 | 11.6 | -59 | -79 | 14.2 | +25 | -4 | -84.0† (-137.0, -31.0) | -75.6 (-153.2, +1.9) |
| Non-HDL-C | 262.9 | -19 | -28 | 271.3 | +5 | +12 | -24.2† (-40.5, -7.9) | -39.7† (-63.1, -16.3) |
Abbreviations: apoB-48 = apolipoprotein B-48; apoC-III = apolipoprotein CIII; non-HDL-C = non-high density lipoprotein cholesterol; N = number of patients; CI = confidence interval; BL = baseline.
Note: Analyses results were based on an analysis of covariance model with treatment, the two randomisation stratification factors, prior history of pancreatitis within 10 years prior to screening (yes vs. no), previous treatment with the unconjugated ASO (yes vs. no) as the fixed effects and log-transformed baseline value as a covariate. Missing data was imputed using placebo washout imputation. The 95% CIs of treatment differences were calculated using a robust variance estimator.
* Reached statistical significance (p value <0.05).
† Reached nominal significance (p value <0.05).
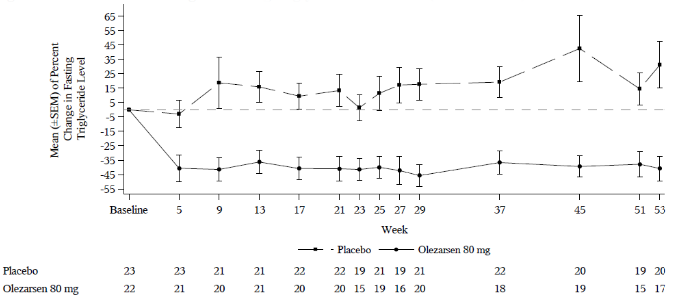
The placebo adjusted percent change in TG levels from baseline at month 12 in the olezarsen 80 mg treated group was nominally significant (Table 2). Following administration of olezarsen 80 mg dose every 4 weeks, a decrease in fasting apoC-III was observed at the first assessment (week 5). The placebo-corrected, percent change from baseline was -57%, -69%, -74%, and -81% at months 1, 3, 6, and 12, respectively. Reductions in apoB-48 and non-HDL-C levels in the olezarsen 80 mg treated group were demonstrated at month 6 and were sustained at month 12. Mean percent changes in TG levels from baseline over time demonstrated a consistent lowering effect during the 12-month treatment period (Figure 1).
Figure 1. Percent change in fasting triglyceride over time (Balance trial):
Over the 12-month treatment period, the numerical incidence of pancreatitis in patients treated with olezarsen 80 mg was lower compared with placebo (1 patient experienced 1 event of adjudicated acute pancreatitis in the olezarsen 80 mg group compared with 11 events experienced by 7 patients in the placebo group). The time to first pancreatitis event was longer in the olezarsen 80 mg group (357 days) compared to placebo (9 days). The mean pancreatitis event rate per 100 patient years was 4.37 for the total olezarsen group (80 mg and 50 mg group) compared with 36.31 for the placebo group. The mean pancreatitis event rate ratio for total olezarsen to placebo was 0.12 (95% CI: 0.022, 0.656).
Elderly population
In clinical trials, 111 (38%) patients treated with olezarsen were ≥65 years of age. No overall differences in safety or efficacy were observed between these patients and younger adult patients.
Immunogenicity
In the Balance trial, with duration of treatment up to 53 weeks, anti-drug antibodies (ADA) were very commonly detected, with 18 out of 43 (42%) patients treated with olezarsen developing treatment-emergent ADAs. No evidence of ADA impact on pharmacodynamics, safety, or efficacy was observed; however, data are limited.
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with olezarsen in one or more subsets of the paediatric population in the treatment of FCS (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
The pharmacokinetic (PK) properties of olezarsen were evaluated following subcutaneous administration of single and multiple doses (once every week, and once every 4 weeks) in healthy subjects and multiple doses (once every 4 weeks) in patients with FCS.
Olezarsen maximum concentration (Cmax) and area under the curve (AUC) showed a slightly greater than dose-proportional increase following single subcutaneous doses ranging from 10 to 120 mg (i.e. 0.13- to 1.5- times the recommended dose) in healthy volunteers.
Population estimates (mean ± SD) of steady state Cmax, and AUC over the dosing interval (AUCτ) were 883 ± 662 ng /mL and 7 440 ± 3 880 ng*h/mL, respectively, following 80 mg monthly dosing in patients with FCS. No accumulation of olezarsen Cmax and AUC was observed after repeated dosing (once every 4 weeks).
Absorption
Following subcutaneous administration, olezarsen is rapidly absorbed with the time to maximum plasma concentration of approximately 2 hours post dose, based on population estimates.
Distribution
Olezarsen is expected to distribute primarily to the liver and kidney cortex after subcutaneous dosing. Olezarsen is bound to human plasma proteins (>99%) in vitro. The population estimates for the apparent central volume of distribution is 91.9 L and the apparent peripheral volume of distribution is 2 960 L.
Biotransformation
Olezarsen is not a substrate for CYP metabolism, and is metabolized by endo- and exonucleases to short oligonucleotide fragments of varying sizes.
Elimination
The terminal elimination half-life is approximately 4 weeks.
The mean fraction of unchanged ASO eliminated in urine was less than 1% of the administered dose within 24 hours.
Immunogenicity
Observed incidence of ADA is highly dependent on the sensitivity and specificity of the assay. In the Balance trial, the presence of ADAs did not affect olezarsen plasma Cmax but increased trough concentrations (Ctrough).
Special populations
Renal impairment
No formal clinical trials have been conducted to investigate the effect of renal impairment on olezarsen PK. A population pharmacokinetic and pharmacodynamic analysis showed no clinically meaningful differences 10 in the pharmacokinetics or pharmacodynamics of olezarsen based on mild and moderate renal impairment (eGFR ≥30 to <90 mL/min/1.73 m²).
Olezarsen has not been studied in patients with severe renal impairment or end-stage renal disease.
Hepatic impairment
No formal clinical trials have been conducted to investigate the effect of hepatic impairment on olezarsen PK. A population pharmacokinetic and pharmacodynamic analysis showed no clinically meaningful differences in the pharmacokinetics or pharmacodynamics of olezarsen based on mild hepatic impairment (total bilirubin ≤ ULN with AST > ULN; or total bilirubin > 1-1.5× ULN with any AST).
Olezarsen has not been studied in patients with moderate or severe hepatic impairment.
Age, gender, weight and race
Based on the population pharmacokinetic and pharmacodynamic analysis, body weight (ranging from 45 to 131 kg), gender, and race have no clinically meaningful effect on olezarsen exposure or apoC-III and triglyceride reductions at steady-state.
No overall differences in pharmacokinetics were observed between adult and elderly patients (age ≥65 years).
5.3. Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential, toxicity to reproduction and development.
In animal studies of the unconjugated form of olezarsen, volanesorsen, available data have shown excretion of very low amounts of volanesorsen in milk. Owing to poor oral bioavailability of volanesorsen, it is considered unlikely that these low milk concentrations would result in systemic exposure from nursing.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.