TURALIO Capsule Ref.[10160] Active ingredients:
Source: FDA, National Drug Code (US) Revision Year: 2020
12.1. Mechanism of Action
Pexidartinib is a small molecule tyrosine kinase inhibitor that targets colony stimulating factor 1 receptor (CSF1R), KIT proto-oncogene receptor tyrosine kinase (KIT), and FMS-like tyrosine kinase 3 (FLT3) harboring an internal tandem duplication (ITD) mutation. Overexpression of the CSF1R ligand promotes cell proliferation and accumulation in the synovium. In vitro, pexidartinib inhibited proliferation of cell lines dependent on CSF1R and ligand-induced autophosphorylation of CSF1R. Pexidartinib also inhibited the proliferation of a CSF1R dependent cell line in vivo.
12.2. Pharmacodynamics
Exposure-Response Relationships
There is an exposure response relationship between pexidartinib steady state exposure and serum transaminase levels (ALT and AST) with a higher risk of increased serum transaminases at higher exposure. Additionally, increased transaminases occurred more frequently with higher pexidartinib doses (200 to 1200 mg per day).
Cardiac Electrophysiology
At two times the mean maximum exposure of the 400 mg twice daily dose, TURALIO does not prolong the QTc interval to any clinically relevant extent.
12.3. Pharmacokinetics
The pharmacokinetics of TURALIO was evaluated following single doses in healthy subjects and following multiple doses in patients as summarized in Table 9.
Table 9. TURALIO Exposure and Pharmacokinetic Parameters:
| General Information | ||
|---|---|---|
| Steady state exposure [Mean (SD)]* | Cmax | 8625 (2746) ng/mL |
| AUC0-12h | 77465 (24975) ng∙h/mL | |
| Dose proportionality | Pexidartinib exposure (Cmax and AUC0-INF) increased linearly over the single oral dose range of 200 to 2400 mg (0.5 to 6 times the recommended dose). | |
| Time to steady state† | Approximately 7 days | |
| Accumulation ratio (AUC) [Median]† | 3.6 | |
| Absorption | ||
| Tmax [Median] | 2.5 hours | |
| Effect of food | ||
| Administration with high-fat meal‡ | • Increased pexidartinib Cmax and AUC0-INF by 100% • Delayed Tmax by 2.5 hours | |
| Administration with low-fat meal§ | • Increased Cmax by 56% and AUC0-INF by 59% • Delayed Tmax by 1.5 hours | |
| Distribution | ||
| In vitro plasma protein binding | • Greater than 99% • Human serum albumin: 99.9% • α-1 acid glycoprotein: 89.9% | |
| Apparent volume of distribution (Vz/F) [Mean (CV%)]¶ | • 187 L (27%) | |
| Elimination | ||
| Apparent clearance [Mean (CV%)]¶ | • 5.1 L/h (36%) | |
| t1/2 [Mean (SD)] | • 26.6 (6.5) hours | |
| Metabolism | ||
| Primary pathway | • Oxidation: CYP3A4 • Glucuronidation: UGT1A4 | |
| N-glucuronide metabolite | • Major inactive metabolite formed by UGT1A4 • Approximately 10% higher exposure than pexidartinib after a single dose | |
| Excretion# | ||
| • Feces: 65% (44% as unchanged) • Urine: 27% as metabolites (≥10% as N-glucuronide) | ||
* Pexidartinib 400 mg twice daily
† Estimated based on half life
‡ The high-fat meal comprised 800 to 1000 calories with fat being 50% of total caloric content; approximately 150 calories from protein, 250 calories from carbohydrates, and 500-600 calories from fat.
§ The low-fat meal comprised 387 calories with fat being approximately 25% of total caloric content.
¶ After a single oral dose of pexidartinib 400 mg#After a single oral dose of radiolabeled pexidartinib 400 mg.
Specific Populations
No clinically meaningful differences in the pharmacokinetics of pexidartinib were observed based on age (18 to 84 years), sex, race (White and Black), or mild hepatic impairment (total bilirubin ≤ ULN with AST > ULN or total bilirubin >1 to 1.5 × ULN with any AST).
Patients with Renal Impairment
Mild (CLcr 60 to 89 mL/min), moderate (CLcr 30 to 59 mL/min) and severe (CLcr 15 to 29 mL/min) renal impairment increased pexidartinib exposure (AUC) by approximately 30%, relative to that in patients with normal renal function (CLcr ≥90 mL/min).
Patients with Hepatic Impairment
The pharmacokinetics of pexidartinib has not been adequately characterized in patients with moderate hepatic impairment (total bilirubin >1.5 to 3 × ULN and any AST) or studied in patients with severe hepatic impairment (total bilirubin >3 to 10 × ULN and any AST).
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
Effects of Other Drugs on Pexidartinib:
Strong CYP3A Inducers: Coadministration of rifampicin (strong CYP3A inducer) decreased pexidartinib Cmax by 33% and AUC0-INF by 65%.
Moderate CYP3A Inducers: Coadministration of efavirenz (moderate CYP3A inducer) is predicted to decrease pexidartinib Cmax by 27% and AUC by 38% at steady state.
Strong CYP3A Inhibitors: Coadministration of itraconazole (strong CYP3A inhibitor) increased pexidartinib Cmax by 48% and AUC0-INF by 70%.
Moderate CYP3A Inhibitors: Coadministration of fluconazole (moderate CYP3A inhibitor) is predicted to increase pexidartinib Cmax by 41% and AUC by 67% at steady state.
UGT Inhibitors: Coadministration of probenecid (UGT inhibitor) increased pexidartinib Cmax by 5% and AUC0-INF by 60%.
Acid-Reducing Agents: Coadministration of esomeprazole (proton pump inhibitor) decreased pexidartinib Cmax by 55% and AUC0-INF by 50%. The effects of H2-receptor antagonists and locally-acting antacids on pexidartinib pharmacokinetics have not been studied.
Effects of Pexidartinib on Other Drugs:
CYP2C19 Substrates: Coadministration of a single oral dose of TURALIO 1800 mg (4.5 times the approved recommended dose of 400 mg) decreased omeprazole (CYP2C19 substrate) Cmax by 37% and AUC0-INF by 17%.
CYP3A Substrates:Coadministration of TURALIO 400 mg twice daily decreased midazolam (CYP3A substrate) Cmax by 28% and AUC0-INF by 59%.
CYP2C9 Substrates: Coadministration of TURALIO 400 mg twice daily increased tolbutamide (CYP2C9 substrate) AUC0-INF by 28%. The effect of pexidartinib on CYP2C9 substrates is not considered clinically relevant.
P-gp Substrates: Coadministration of a single oral dose of TURALIO 1800 mg (4.5 times the approved recommended dose of 400 mg) increased digoxin (P-gp substrate) Cmax by 30% and AUC0-INF by 9%.
CYP2C8 Substrates: The effect of pexidartinib on CYP2C8 substrates is predicted not to be clinically relevant.
In Vitro Studies
CYP/UGT Enzymes:
Pexidartinib is likely to inhibit CYP2B6 and induce CYP2B6 at clinically relevant concentrations. Pexidartinib is likely to inhibit UGT1A1 at clinically relevant concentrations.
Transporter Systems:
Pexidartinib is not a substrate for P-gp, BCRP, OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3, OATP2B1, and BSEP.
Pexidartinib is an inhibitor of MATE1, MATE2-K, OATP1B1, OATP1B3 and OATP2B1.
13.1. Carcinogensis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies were performed in mice and rats. Both studies were negative for carcinogenic findings at exposures up to 9 times the human exposure at the recommended daily dose of 800 mg based on AUC.
Pexidartinib was not mutagenic in an in vitro bacterial reverse mutation (AMES) assay or clastogenic in either an in vitro human peripheral blood lymphocyte chromosomal aberrations assay or in an in vivo mouse bone marrow micronucleus assay.
Based on nonclinical findings, TURALIO may impair male and female fertility. In a fertility study in which pexidartinib was administered orally to male and female rats, there were reductions in pregnancy, as well as increases in pre- and post-implantation loss with a corresponding reduction in viable embryos at 40 mg/kg (approximately 1.3 times the human exposure at the recommended dose of 800 mg). Males at this dose level displayed reductions in spermatogenic parameters and adverse effects on sperm concentration, production, motility, and morphology. Lower testicular and epididymal weights occurred in this study at doses of ≥10 mg/kg/day (approximately 0.3 times the human exposure at the recommended dose of 800 mg). This is consistent with findings in chronic toxicology studies of germ cell depletion of the testes and hypospermia and cellular debris in the epididymis in male reproductive tissues of both rats and dogs at respective doses as low as 20 and 30 mg/kg/day (approximately 0.6 and 0.1 times the human exposure at the recommended dose of 800 mg). In rats, these changes persisted following a 16-week recovery period at the 60 mg/kg/day dose level (approximately 1.5 times the human exposure at the recommended dose of 800 mg).
In female rats, necrosis of corpora lutea occurred at doses ≥0.5 mg/kg/day (approximately 0.01 times the human exposure at the recommended dose of 800 mg) with pigment deposition within the interstitium of the ovaries, an increased incidence of luteal cysts and incidence/severity of hemorrhage of corpora lutea, and a decreased incidence of retained antral follicles and decreased corpora lutea at 60 mg/kg (approximately 1.8 times the human exposure at the recommended dose of 800 mg). In female dogs there were decreased follicle numbers and moderate atrophy of the oviduct, uterus, and cervix at doses as low as 1 mg/kg (approximately 0.01 times the human exposure at the recommended dose of 800 mg).
13.2. Animal Toxicology and/or Pharmacology
In repeat dose toxicity studies of up to 26 weeks in rats, there were findings of myxomatous change in the skin, tongue, and gastrointestinal tract, lymphoid depletion of the bone marrow and thymus, and chronic progressive nephropathy of the kidney at 20 mg/kg/day (approximately 0.6 times the human exposure at the recommended dose of 800 mg). Similar changes occurred in the rat carcinogenicity study along with alterations in the tunica intima of the aorta. Vascular inflammation consistent with polyarteritis nodosa occurred in male rats at 60 mg/kg/day (approximately 1.5 times the human exposure at the recommended dose of 800 mg). There were also dose-dependent findings of minimal to moderate subphyseal or cortical hyperostosis and physeal hypertrophy in the femur that correlated with decreased systemic phosphate levels at doses ≥60 mg/kg.
14. Clinical Studies
14.1 Tenosynovial Giant Cell Tumor
The efficacy of TURALIO was evaluated in ENLIVEN (NCT02371369), a double-blind, randomized (1:1), placebo-controlled, multicenter trial in patients with symptomatic TGCT [also referred to as giant cell tumor of the tendon sheath (GCT-TS) or pigmented villonodular synovitis (PVNS)] for whom surgical removal of the tumor would be associated with worsening functional limitation or severe morbidity. Eligible patients were required to have measurable disease per the Response Evaluation Criteria in Solid Tumors (RECIST) v1.1. Patients were randomized to placebo or TURALIO 400 mg in the morning and 600 mg in the evening for 2 weeks followed by 400 mg twice daily. Treatment continued until unacceptable toxicity or disease progression. Randomization was stratified by geographic region (US vs. non-US countries) and disease location (upper extremity vs. lower extremity involvement).
The major efficacy outcome measure was overall response rate (ORR) as assessed by blinded independent central review (BICR) at Week 25 using RECIST v1.1. Additional efficacy outcome measures were mean change from baseline in range of motion of the affected joint at Week 25 and ORR as assessed by BICR at Week 25 using tumor volume score (TVS). Range of motion was measured as a percent of normal reference range for the affected joint. Range of motion assessments were performed by a third-party clinical assessor using a goniometer. TVS was defined in ENLIVEN as the estimated volume of the maximally distended synovial cavity or tendon sheath involved, measured in 10% increments. Patients in the placebo arm were offered TURALIO at Week 25 beginning with a 400 mg twice daily dose, as permitted by the study protocol.
A total of 120 patients were randomized, 61 to the TURALIO arm and 59 to the placebo arm. The median age was 44 years (range: 18-79); 59% were females; 88% were White; 53% had prior surgery; 88% were diagnosed with diffuse TGCT; and 9% had previously been treated with systemic therapy. Disease locations were knee (61%), ankle (18%), hip (11%), wrist (3%), foot (3%) and other (5%).
ENLIVEN demonstrated a statistically significant improvement in ORR in patients randomized to TURALIO compared with placebo. Efficacy results are summarized in Table 10.
Table 10. Efficacy Results Assessed at Week 25 for ENLIVEN:
| Efficacy Parameter | TURALIO N=61 | Placebo N=59 |
|---|---|---|
| Overall Response Rate (ORR)*,† | ||
| ORR (95% CI) | 38% (27%, 50%) | 0 (0, 6%) |
| Complete Response | 15% | 0 |
| Partial Response | 23% | 0 |
| P-value‡ | <0.0001 | |
| Duration of Response (DOR)† | ||
| Range (months) | 6.9+, 24.9+ | NA |
CI: confidence interval; NA: not applicable; SD: standard deviation; LS: least squares; +: denotes ongoing at last assessment
* Blinded independent central review
† Data cut-off date January 31, 2018
‡ Fisher's exact test
The analysis of mean change from baseline in range of motion at Week 25 demonstrated a statistically significant improvement in patients randomized to TURALIO compared to placebo. Figure 1 shows the change from baseline in range of motion for each patient at Week 25 (TURALIO N=45, placebo N=43). Results were excluded for 1 patient with missing baseline and 31 patients with a missing range of motion assessment at Week 25.
Figure 1. Change from Baseline in Range of Motion at Week 25 for ENLIVEN:
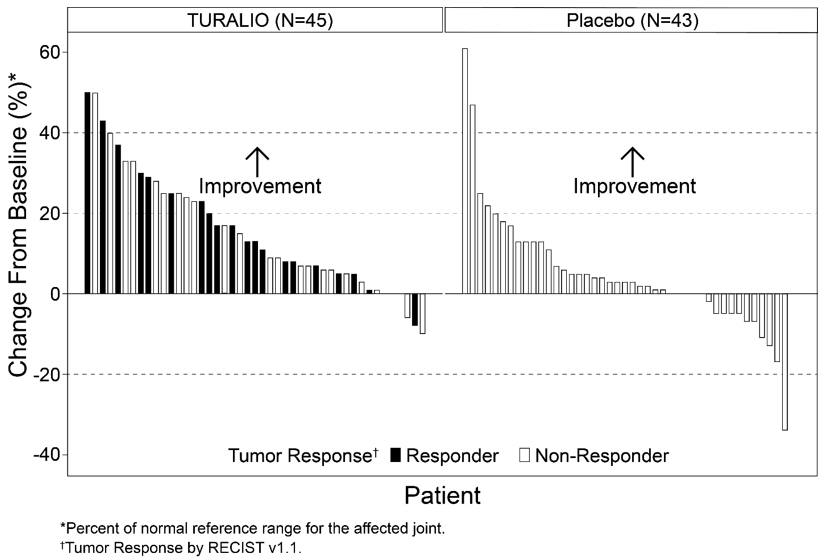
ORR by TVS was 56% (95% CI: 43%, 67%) in patients randomized to the TURALIO arm and 0% in patients randomized to the placebo arm; p<0.0001.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.