UBRELVY Tablet Ref.[10378] Active ingredients:
Source: FDA, National Drug Code (US) Revision Year: 2020
12.1. Mechanism of Action
Ubrogepant is a calcitonin gene-related peptide receptor antagonist.
12.2. Pharmacodynamics
Cardiac Electrophysiology
At a dose 2 times the maximum recommended daily dose, UBRELVY does not prolong the QT interval to any clinically relevant extent.
12.3. Pharmacokinetics
Absorption
Following oral administration of UBRELVY, ubrogepant is absorbed with peak plasma concentrations at approximately 1.5 hours. Ubrogepant displays dose-proportional pharmacokinetics within the recommended dose range [see Dosage and Administration (2.1)].
Effect of Food
When UBRELVY was administered with a high-fat meal, the time to maximum ubrogepant plasma concentration was delayed by 2 hours and resulted in a 22% reduction in Cmax with no change in AUC. UBRELVY was administered without regard to food in clinical efficacy studies [see Dosage and Administration (2.1)].
Distribution
Plasma protein binding of ubrogepant is 87% in vitro. The mean apparent central volume of distribution of ubrogepant (V/F) after single dose oral administration is approximately 350 L.
Elimination
Metabolism
Ubrogepant is eliminated mainly through metabolism, primarily by CYP3A4. The parent compound (ubrogepant), and 2 glucuronide conjugate metabolites were the most prevalent circulating components in human plasma. The glucuronide metabolites are not expected to contribute to the pharmacological activity of ubrogepant since they were reported as about 6000-fold less potent in the CGRP receptor binding assay.
Excretion
The elimination half-life of ubrogepant is approximately 5-7 hours. The mean apparent oral clearance (CL/F) of ubrogepant is approximately 87 L/hr. Ubrogepant is excreted mostly via the biliary/fecal route, while the renal route is a minor route of elimination. Following single oral dose administration of [14C]-ubrogepant to healthy male subjects, 42% and 6% of the dose was recovered as unchanged ubrogepant in feces and urine, respectively.
Specific Populations
Patients with Renal Impairment
Population pharmacokinetic analysis based on pooled data from clinical studies was used to evaluate the effect of renal impairment characterized based on estimated creatinine clearance (CLcr) using the Cockcroft-Gault (C-G) equation. Renal impairment did not reveal a significant difference in the pharmacokinetics of ubrogepant in patients with mild or moderate renal impairment (CLcr 30-89 mL/min) relative to those with normal renal function (CLcr >90 mL/min). Patients with severe renal impairment or ESRD (eGFR <30 mL/min) have not been studied. Dose adjustment in patients with severe renal impairment (CLcr 15-29 mL/min) is recommended based on ADME information and a conservative assumption that severe renal impairment is unlikely to cause more than a two-fold increase in exposure of ubrogepant [see Dosage and Administration (2.2)]. No dosing recommendations can be made for patients with ESRD (CLcr<15 mL/min).
Patients with Hepatic Impairment
In patients with pre-existing mild (Child-Pugh Class A), moderate (Child-Pugh Class B), or severe hepatic impairment (Child-Pugh Class C), ubrogepant exposure was increased by 7%, 50%, and 115%, respectively. Patients with severe hepatic impairment require dose adjustments [see Dosage and Administration (2.2)].
Other Specific Populations
Based on a population pharmacokinetic analysis, age, sex, race, and body weight did not have a significant effect on the pharmacokinetics (Cmax and AUC) of ubrogepant. Therefore, no dose adjustments are warranted based on these factors.
Drug Interactions
In Vitro Studies
Enzymes:
Ubrogepant is not an inhibitor of CYP1A2, 2B6, or 3A4. Ubrogepant is a weak inhibitor of CYP2C8, 2C9, 2D6, 2C19, MAO-A, and UGT1A1. The in vitro inhibition potential is not expected to be clinically significant. Ubrogepant is not an inducer of CYP1A2, 2B6, or 3A4 at clinically relevant concentrations.
Transporters:
Ubrogepant is a substrate of BCRP and P-gp transporters in-vitro; therefore, use of inhibitors of BCRP and/or P-gp may increase the exposure of ubrogepant. Dose adjustment for concomitant use of UBRELVY with inhibitors of BCRP and/or P-gp is recommended based on ADME and clinical interaction studies with CYP3A4/P-gp inhibitors that show the highest predicted potential increase in exposure of ubrogepant is not expected to be more than two-fold [see Dosage and Administration (2.2) and Drug Interactions (7.3)].
Ubrogepant is a weak substrate of OATP1B1, OATP1B3, and OAT1, but not a substrate of OAT3. It is not an inhibitor of P-gp, BCRP, BSEP, MRP3, MRP4, OAT1, OAT3, or NTCP transporters, but is a weak inhibitor of OATP1B1, OATP1B3, and OCT2 transporters. Dose adjustments are needed only for P-gp, or BCRP inhibitors. No clinical drug interactions are expected for UBRELVY with other transporters.
In Vivo Studies
CYP3A4 Inhibitors [see Dosage and Administration (2.2), Contraindications (4), and Drug Interactions (7.1)]:
Co-administration of UBRELVY with ketoconazole, a strong CYP3A4 inhibitor, resulted in a 9.7-fold and 5.3-fold increase in AUCinf and Cmax of ubrogepant, respectively. Co-administration of UBRELVY with verapamil, a moderate CYP3A4 inhibitor, resulted in about 3.5-fold and 2.8-fold increase in AUCinf and Cmax of ubrogepant, respectively. No dedicated drug interaction study was conducted to assess concomitant use with weak CYP3A4 inhibitors. The conservative prediction of the maximal potential increase in ubrogepant exposure with weak CYP3A4 inhibitors is not expected to be more than 2-fold.
CYP3A4 Inducers [see Dosage and Administration (2.2) and Drug Interactions (7.2)]:
Co-administration of UBRELVY with rifampin, a strong CYP3A4 inducer, resulted in an 80% reduction in ubrogepant exposure. No dedicated drug interaction studies were conducted to assess concomitant use with weak or moderate CYP3A4 inducers. Dose adjustment for concomitant use of UBRELVY with weak or moderate CYP3A4 inducers is recommended based on a conservative prediction of 50% reduction in exposure of ubrogepant.
Other Drug-Drug Interaction Evaluations:
No significant pharmacokinetic interactions were observed for either ubrogepant or co-administered drugs when UBRELVY was administered with oral contraceptives (containing norgestimate and ethinyl estradiol), acetaminophen, naproxen, sumatriptan, or esomeprazole (a proton pump inhibitor).
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity
Two-year oral carcinogenicity studies of ubrogepant were conducted in mice (0, 5, 15, or 50 mg/kg/day) and rats (0, 10, 30, or 100 mg/kg in males; 0, 10, 30, or 150 mg/kg in females). There was no evidence of drug-related tumors in either species. The highest dose tested in mice is similar to the maximum recommended human dose (200 mg/day) on a body surface area (mg/m²) basis. Plasma exposure (AUC) at the highest dose tested in rats is approximately 25 times that in humans at the maximum recommended human dose (MRHD) of 200 mg/day.
Mutagenicity
Ubrogepant was negative in in vitro (Ames, chromosomal aberration test in Chinese Hamster Ovary cells) and in vivo (rat bone marrow micronucleus) assays.
Impairment of Fertility
Oral administration of ubrogepant (0, 20, 80, or 160 mg/kg/day) to male and female rats (mated with drug-naïve females and males, respectively) resulted in no adverse effects on fertility or reproductive performance. Plasma exposures (AUC) at the highest dose tested are approximately 30 times that in humans at the MRHD.
14. Clinical Studies
The efficacy of UBRELVY for the acute treatment of migraine was demonstrated in two randomized, double-blind, placebo-controlled trials [Study 1 (NCT02828020) and Study 2 (NCT02867709)]. Study 1 randomized patients to placebo (n=559) or UBRELVY 50 mg (n=556) or 100 mg (n=557) and Study 2 randomized patients to placebo (n=563) or UBRELVY 50 mg (n=562). In all studies, patients were instructed to treat a migraine with moderate to severe headache pain intensity. A second dose of study medication (UBRELVY or placebo), or the patient's usual acute treatment for migraine, was allowed between 2 to 48 hours after the initial treatment for a non-responding or recurrent migraine headache. Up to 23% of patients were taking preventive medications for migraine at baseline. None of these patients were on concomitant preventive medication that act on the CGRP pathway.
The primary efficacy analyses were conducted in patients who treated a migraine with moderate to severe pain. The efficacy of UBRELVY was established by an effect on pain freedom at 2 hours post-dose and most bothersome symptom (MBS) freedom at 2 hours post-dose, compared to placebo, for Studies 1 and 2. Pain freedom was defined as a reduction of moderate or severe headache pain to no pain, and MBS freedom was defined as the absence of the self-identified MBS (i.e., photophobia, phonophobia, or nausea). Among patients who selected an MBS, the most commonly selected was photophobia (56%), followed by phonophobia (24%), and nausea (19%).
In both studies, the percentage of patients achieving headache pain freedom and MBS freedom 2 hours post-dose was significantly greater among patients receiving UBRELVY compared to those receiving placebo (see Table 3). Table 3 also presents the results of the analyses of the percentage of patients achieving pain relief at 2 hours (defined as a reduction in migraine pain from moderate or severe to mild or none) post-dose and the percentage of patients achieving sustained pain freedom between 2 to 24 hours post-dose.
The incidence of photophobia and phonophobia was reduced following administration of UBRELVY at both doses (50 mg and 100 mg) as compared to placebo.
Table 3. Migraine Efficacy Endpoints for Study 1 and Study 2:
| Study 1 | Study 2 | ||||
|---|---|---|---|---|---|
| UBRELVY 50 mg | UBRELVY 100 mg | Placebo | UBRELVY 50 mg | Placebo | |
| Pain Free at 2 hours | |||||
| N | 422 | 448 | 456 | 464 | 456 |
| % Responders | 19.2 | 21.2 | 11.8 | 21.8 | 14.3 |
| Difference from placebo (%) | 7.4 | 9.4 | 7.5 | ||
| p value | 0.002 | <0.001 | 0.007 | ||
| Most Bothersome Symptom Free at 2 hours | |||||
| N | 420 | 448 | 454 | 463 | 456 |
| % Responders | 38.6 | 37.7 | 27.8 | 38.9 | 27.4 |
| Difference from placebo (%) | 10.8 | 9.9 | 11.5 | ||
| p value | <0.001 | <0.001 | <0.001 | ||
| Pain Relief at 2 hours | |||||
| N | 422 | 448 | 456 | 464 | 456 |
| % Responders | 60.7 | 61.4 | 49.1 | 62.7 | 48.2 |
| p value | <0.001 | <0.001 | <0.001 | ||
| Sustained Pain Freedom 2-24 hours | |||||
| N | 418 | 441 | 452 | 457 | 451 |
| % Responders | 12.7 | 15.4 | 8.6 | 14.4 | 8.2 |
| p value | *NS | 0.002 | 0.005 | ||
* Not statistically significant (NS)
Figure 1 presents the percentage of patients achieving migraine pain freedom within 2 hours following treatment in Studies 1 and 2.
Figure 1. Percentage of Patients Achieving Pain Freedom within 2 Hours in Pooled Studies 1 and 2:
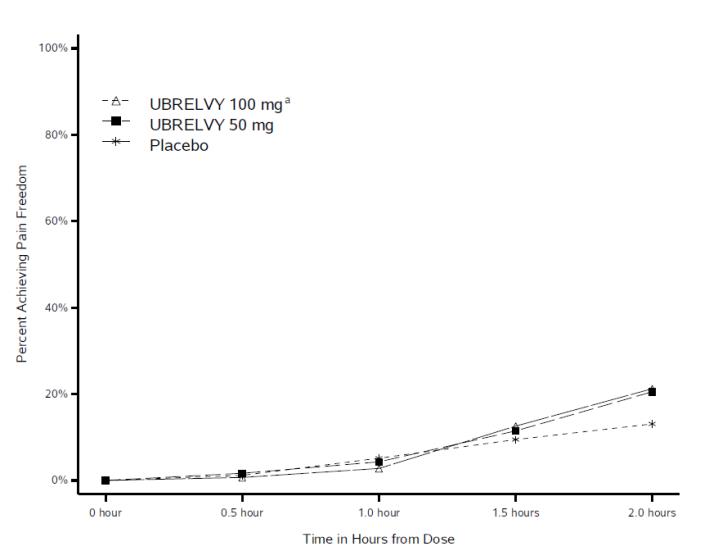
a The 100 mg arm was only included in Study 1.
Figure 2 presents the percentage of patients achieving MBS freedom within 2 hours in Studies 1 and 2.
Figure 2. Percentage of Patients Achieving MBS Freedom within 2 Hours in Pooled Studies 1 and 2:
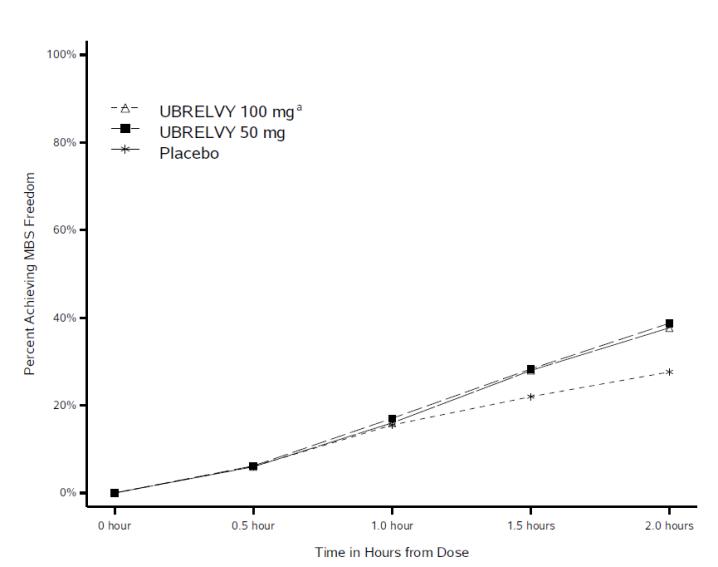
a The 100 mg arm was only included in Study 1.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.