UNITUXIN Solution for injection Ref.[10022] Active ingredients: Dinutuximab
Source: FDA, National Drug Code (US) Revision Year: 2020
12.1. Mechanism of Action
Dinutuximab binds to the glycolipid GD2. This glycolipid is expressed on neuroblastoma cells and on normal cells of neuroectodermal origin, including the central nervous system and peripheral nerves. Dinutuximab binds to cell surface GD2 and induces cell lysis of GD2-expressing cells through antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC).
12.3. Pharmacokinetics
The pharmacokinetics of dinutuximab was evaluated by a population pharmacokinetic analysis in a clinical study of Unituxin in combination with GM-CSF, IL-2, and RA. In this study, 27 children with high-risk neuroblastoma (age: 3.9±1.9 years) received up to 5 cycles of Unituxin at 17.5 mg/m²/day as an intravenous infusion over 10 to 20 hours for 4 consecutive days every 28 days. The observed maximum plasma dinutuximab concentration (Cmax) was 11.5 mcg/mL (20%, coefficient of variation [CV]). The mean volume of distribution at steady state (Vdss) was 5.4 L (28%). The clearance was 0.21 L/day (62%) and increased with body size. The terminal half-life was 10 days (56%).
No formal pharmacokinetic studies were conducted in patients with renal or hepatic impairment.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
No animal studies have been conducted to evaluate the carcinogenic or mutagenic potential of dinutuximab.
Dedicated studies examining the effects of dinutuximab on fertility in animals have not been conducted. No clear effects on reproductive organs were observed in general toxicology studies conducted in rats.
13.2. Animal Toxicology and/or Pharmacology
Nonclinical studies suggest that dinutuximab-induced neuropathic pain and neurotoxicity are mediated by binding of the antibody to the GD2 antigen located on the surface of nerve fibers and myelin and subsequent induction of CDC and ADCC activity.
14. Clinical Studies
The safety and effectiveness of Unituxin was evaluated in a randomized, open-label, multicenter trial conducted in pediatric patients with high-risk neuroblastoma (Study 1). All patients had received prior therapy consisting of induction combination chemotherapy, maximum feasible surgical resection, myeloablative consolidation chemotherapy followed by autologous stem cell transplant, and radiation therapy to residual soft tissue disease. Patients were randomized between Day 50 and Day 77 post-autologous stem cell transplantation.
Patients were required to have achieved at least a partial response prior to autologous stem cell transplantation, have no evidence of disease progression following completion of front-line multi-modality therapy, have adequate pulmonary function (no dyspnea at rest and peripheral arterial oxygen saturation of at least 94% on room air), adequate hepatic function (total bilirubin <1.5× the upper limit of normal and ALT <5× the upper limit of normal), adequate cardiac function (shortening fraction of >30% by echocardiogram, or if shortening fraction abnormal, ejection fraction of 55% by gated radionuclide study), and adequate renal function (glomerular filtration rate at least 70 mL/min/1.73 m²). Patients with systemic infections or a requirement for concomitant systemic corticosteroids or immunosuppressant usage were not eligible for enrollment.
Patients randomized to the Unituxin/RA arm received up to 5 cycles of Unituxin (clinical trials material) in combination with granulocyte-macrophage colony-stimulating factor (GM-CSF) (Table 8) or interleukin-2 (IL-2) (Table 9) plus 13-cis-retinoic acid (RA), followed by 1 cycle of RA alone. Patients randomized to the RA arm received 6 cycles of RA. Unituxin was administered at a dose of 17.5 mg/m²/day (equivalent to 25 mg/m²/day of clinical trials material) on 4 consecutive days. Patients in both treatment arms received 6 cycles of RA at a dose of 160 mg/m²/day orally (for patients weighing more than 12 kg) or 5.33 mg/kg/day (for patients weighing less than or equal to 12 kg) in 2 divided doses for 14 consecutive days.
Table 8. Dosage Regimen in the Unituxin/RA Arm for Cycles 1, 3, and 5:
| Cycle Day | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15-24 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| GM-CSF* | X | X | X | X | X | X | X | X | X | X | X | X | X | X | |
| Unituxin† | X | X | X | X | |||||||||||
| RA‡ | X | X | X | X | X |
* GM-CSF: 250 µg/m²/day, administered by either subcutaneous injection (recommended) or IV infusion administered over 2 hours.
† Unituxin: 17.5 mg/m²/day, administered by diluted IV infusion over 10 to 20 hours.
‡ RA: for >12 kg body weight, 80 mg/m orally twice daily for a total dose of 160 mg/m²/day; for ≤12 kg body weight, 2.67 mg/kg orally twice daily for a total daily dose of 5.33 mg/kg/day (round dose up to nearest 10 mg).
Table 9. Dosage Regimen in the Unituxin/RA Arm for Cycles 2 and 4:
| Cycle Day | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12-14 | 15-28 | 29-32 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IL-2* | X | X | X | X | X | X | X | X | ||||||
| Unituxin† | X | X | X | X | ||||||||||
| RA‡ | X |
* IL-2: 3 MIU/m²/day administered by continuous IV infusion over 96 hours on Days 1 to 4 and 4.5 MIU/m²/day on Days 8 to 11.
† Unituxin: 17.5 mg/m²/day, administered by diluted IV infusion over 10 to 20 hours.
‡ RA: for >12 kg body weight, 80 mg/m orally twice daily for a total dose of 160 mg/m²/day; for ≤12 kg body weight, 2.67 mg/kg orally twice daily for a total daily dose of 5.33 mg/kg/day (round dose up to nearest 10 mg).
A total of 226 patients were randomized, 113 patients to each arm. In general, demographic and baseline tumor characteristics were similar across study arms. Across the study population, 60% were male, the median age was 3.8 years and 3% of patients were less than 1.5 years, 82% were White and 7% were Black. The majority (80%) of patients had International Neuroblastoma Staging System Stage 4 disease. Thirty-five percent of patients had a complete response, 43% had a very good partial response, and 23% had a partial response to therapy received prior to autologous stem cell transplant. Forty-six percent of patients had neuroblastoma that was not MYCN-amplified, 36% had tumors with known MYCN-amplification, and MYCN status was unknown or missing in 19% of patients. Forty-three percent of patients had hyperdiploid tumors, 36% had diploid tumors, and DNA ploidy status was unknown or missing in 21% of patients.
The major efficacy outcome measure was investigator-assessed event-free survival (EFS), defined as the time from randomization to the first occurrence of relapse, progressive disease, secondary malignancy, or death. Overall survival (OS) was also evaluated. After observing a numerical improvement in EFS based on the seventh interim analysis, the Data Monitoring Committee recommended termination of accrual. Efficacy results are shown in Table 10.
Table 10. Efficacy Results:
| Efficacy Parameter | Unituxin/RA arm n=113 | RA arm n=113 | |
|---|---|---|---|
| EFS | No. of Events (%) | 33 (29%) | 50 (44%) |
| Median (95% CI) (years) | NR (3.4, NR) | 1.9 (1.3, NR) | |
| Hazard Ratio (95% CI) | 0.57 (0.37, 0.89) | ||
| p-value (log-rank test)* | 0.01 | ||
| OS† | No. of Events (%) | 31 (27%) | 48 (42%) |
| Median (95% CI) (years) | NR (7.5, NR) | NR (3.9, NR) | |
| Hazard Ratio (95% CI) | 0.58 (0.37, 0.91) | ||
NR, not reached
* Compared to the allocated alpha of 0.01 pre-specified for the seventh interim analysis of EFS
† Based on an additional 3 years of follow up after the seventh interim analysis of EFS
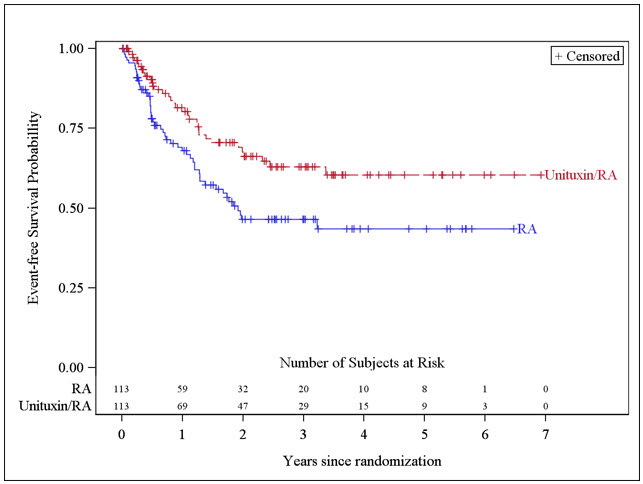
The Kaplan-Meier curve of EFS is shown in Figure 1.
Figure 1. Kaplan-Meier Curve of Event-Free Survival:
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.