UPLIZNA Concentrate for solution for infusion Ref.[109666] Active ingredients: Inebilizumab
Source: European Medicines Agency (EU) Revision Year: 2026 Publisher: Amgen Europe B.V., Minervum 7061, 4817 ZK Breda, The Netherlands
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: immunosuppressants, monoclonal antibodies
ATC code: L04AG10
Mechanism of action
Inebilizumab is a monoclonal antibody that specifically binds to CD19, a cell surface antigen present on pre-B and mature B-cell lymphocytes, including plasmablasts and some plasma cells. Following cell surface binding to B lymphocytes, inebilizumab supports antibody-dependent cellular cytolysis (ADCC) and antibody-dependent cellular phagocytosis (ADCP). B cells are believed to play a central role in the pathogenesis of NMOSD, IgG4-RD and gMG. The precise mechanism by which inebilizumab exerts its therapeutic effects in these diseases is presumed to involve B-cell depletion and may include the suppression of antibody secretion, antigen presentation, B-cell–T-cell interaction, and the production of inflammatory mediators.
Pharmacodynamic effects
Pharmacodynamics of inebilizumab were assessed with an assay for CD20+ B cells, since inebilizumab can interfere with the CD19+ B-cell assay. Treatment with inebilizumab reduces CD20+ B-cell counts in blood by 8 days after infusion. In the clinical study of 174-NMOSD patients, CD20+ B-cell counts were reduced below the lower limit of normal by 4 weeks in 100% of patients treated with inebilizumab and remained below the lower limit of normal in 94% of patients for 28 weeks after initiation of treatment. In the clinical study of 68-IgG4-RD patients, CD20+ B-cell counts were reduced below the lower limit of normal by week 2 in 100% of patients treated with inebilizumab and remained below the lower limit of normal in 82% and 79% of patients at week 26 and 52, respectively, with 6-month treatment interval. In the gMG study, CD20+ B-cell counts were reduced below the lower limit of normal by week 4 in ≥99% of inebilizumab-treated patients in the overall population (anti-AChR-Ab+ and anti-MuSK-Ab+). B cell counts remained below the lower limit of normal in ≥96% of patients in the overall population at the end of the 6-month randomised-controlled period (RCP). After 1 year of inebilizumab treatment in patients with anti-AChR-Ab+ gMG, 92% of patients had B-cell counts below the lower limit of normal. The time to B-cell repletion following administration of inebilizumab is not known.
ADA (anti-drug antibodies)-positive status appeared to have no clinically relevant impact on PK and PD (B-cell) parameters and did not impact the long-term safety profile. There was no apparent effect of ADA status on the efficacy outcome; however, the impact cannot be fully assessed given the low incidence of ADA associated with inebilizumab treatment.
Clinical efficacy and safety
Neuromyelitis optica spectrum disorders (NMOSD)
The efficacy of inebilizumab for the treatment of NMOSD was studied in a randomised (3:1), double-blind, placebo-controlled clinical trial in adults with AQP4-IgG seropositive or seronegative NMOSD. The study included patients who had experienced at least one acute NMOSD attack in the prior year or at least 2 attacks in the prior 2 years that required rescue therapy (e.g., steroids, plasma exchange, intravenous immunoglobulin), and had an Expanded Disability Severity Scale (EDSS) score ≤7.5 (patients with a score of 8.0 were eligible if the patient was reasonably able to participate). Patients were excluded if previously treated with immunosuppressant therapies within an interval specified for each such therapy. Background immunosuppressant therapies for the prevention of NMOSD attacks were not permitted. A 2-week course of oral corticosteroids (plus a 1-week taper) was administered at the start of inebilizumab treatment in the pivotal study.
Patients were treated with intravenous infusions of inebilizumab 300 mg on Day 1 and on Day 15, or matching placebo, and then followed for a period of up to 197 days or an adjudicated attack, termed the randomised-controlled period (RCP). All potential attacks were evaluated by a blinded, independent, Adjudication Committee (AC), who determined whether the attack met protocol-defined criteria. The attack criteria recognised attacks in all domains affected by NMOSD (optic neuritis, myelitis, brain, and brainstem) and included criteria based exclusively on substantial clinical manifestations, as well as criteria that augmented more modest clinical findings with the use of MRI (see Table 3).
Table 3. Overview of the protocol-defined criteria for an NMOSD attack:
| Domain | Representative symptoms | Clinical-only findings | Clinical PLUS radiological findings |
|---|---|---|---|
| Optic nerve | Blurred vision Loss of vision Eye pain | 8 criteria based on changes in visual acuity or relative afferent pupillary defect (RAPD) | 3 criteria based on changes in visual acuity or RAPD plus presence of corresponding optic nerve MRI findings |
| Spinal cord | Deep or radicular pain Extremity paraesthesia Weakness Sphincter dysfunction Lhermitte's sign (not in isolation) | 2 criteria based on changes in pyramidal, bladder/bowel, or sensory functional scores | 2 criteria based on changes in pyramidal, bladder/bowel, or sensory functional scores PLUS corresponding spinal cord MRI findings |
| Brainstem | Nausea Intractable vomiting Intractable hiccups Other neurological signs (e.g., double vision, dysarthria, dysphagia, vertigo, oculomotor palsy, weakness, nystagmus, other cranial nerve abnormality) | None | 2 criteria based on symptoms or changes in brainstem/cerebellar functional scores PLUS corresponding brainstem MRI findings |
| Brain | Encephalopathy Hypothalamic dysfunction | None | 1 criterion based on changes in cerebral/sensory/pyramidal functional scores PLUS corresponding brain MRI findings |
Patients who experienced an AC-determined attack in the RCP, or who completed the Day 197 visit without an attack, exited the RCP and had the option to enrol into an OLP and initiate or continue treatment with inebilizumab.
A total of 230 patients were enrolled: 213 patients were AQP4-IgG seropositive patients and 17 were seronegative patients were enrolled; 174 patients were treated with inebilizumab and 56 patients were treated with placebo in the RCP of the study. Of the 213 AQP4-IgG seropositive patients, 161 were treated with inebilizumab and 52 were treated with placebo in the RCP of the study. Baseline and efficacy results are presented for the AQP4-IgG seropositive patients.
Baseline demographics and disease characteristics were balanced across the 2 treatment groups (see Table 4).
Table 4. Demographics and baseline characteristics of the AQP4-IgG seropositive NMOSD patients:
| Characteristic | Placebo N=52 | Inebilizumab N=161 | Overall N=213 |
|---|---|---|---|
| Age (years): mean (standard deviation [SD]) | 42.4 (14.3) | 43.2 (11.6) | 43.0 (12.3) |
| Age ≥65 years, n (%) | 4 (7.7) | 6 (3.7) | 10 (4.7) |
| Sex: Male, n (%) | 3 (5.8) | 10 (6.2) | 13 (6.1) |
| Sex: Female, n (%) | 49 (94.2) | 151 (93.8) | 200 (93.9) |
| Expanded disability status scale (EDSS): mean (SD) | 4.35 (1.63) | 3.81 (1.77) | 3.94 (1.75) |
| Disease duration (years): mean (SD) | 2.92 (3.54) | 2.49 (3.39) | 2.59 (3.42) |
| Number of prior relapses: ≥2, n (%) | 39 (75.0) | 137 (85.1) | 176 (82.6) |
| Annualised Relapse Rate: mean (SD) | 1.456 (1.360) | 1.682 (1.490) | 1.627 (1.459) |
Rescue therapy was initiated as needed for NMOSD attacks. All patients were pre-medicated prior to investigational product administration to reduce the risk of infusion-related reactions.
The primary efficacy endpoint was time (days) from Day 1 to onset of an AC-determined NMOSD attack on or before Day 197. Additional key secondary endpoint measures included worsening from baseline in EDSS at last visit during the RCP, change from baseline in low-contrast visual acuity binocular score measured by low-contrast Landolt C Broken Rings Chart at last visit during the RCP, cumulative total active MRI lesions (new gadolinium-enhancing or new/enlarging T2 lesions) during the RCP, and the number of NMOSD-related in-patient hospitalisations. A patient was considered to have a worsening in EDSS score if one of the following criteria was met: (1) worsening of 2 or more points in EDSS score for patients with baseline score of 0; (2) worsening of 1 or more points in EDSS score for patients with baseline score of 1 to 5; (3) worsening of 0.5 points or more in EDSS score for patients with baseline score of 5.5 or more. Although no comparator was available during the OLP, the annualised attack rate across both randomised and open-label treatment was determined.
Results in AQP4-IgG seropositive patients are presented in Table 5 and Figure 1. In this study, treatment with inebilizumab statistically significantly reduced the risk of an AC-determined NMOSD attack as compared to treatment with placebo (hazard ratio: 0.227, p<0.0001; 77.3% reduction in risk of AC-determined NMOSD attack) in AQP4-IgG seropositive patients. There was no treatment benefit observed in AQP4-IgG seronegative patients.
In the inebilizumab group EDDS worsening was significantly less than placebo group (14.9% versus 34.6% of the subjects). There were no differences in the low-contrast visual acuity binocular score between the study arms. The mean cumulative number of total active MRI lesions (1.7 versus 2.3) and mean cumulative number of NMOSD related hospitalisations (1.0 versus 1.4) were reduced in the inebilizumab study group.
Table 5. Efficacy results in pivotal trial in AQP4-IgG seropositive NMOSD:
| Treatment group | ||
|---|---|---|
| Placebo N=52 | Inebilizumab N=161 | |
| Time to adjudication committee-determined attack (primary efficacy endpoint) | ||
| Number (%) of patients with attack | 22 (42.3%) | 18 (11.2%) |
| Hazard ratio (95% CI)a | 0.227 (0.1214, 0.4232) | |
| p-valuea | <0.0001 | |
a Cox regression method, with Placebo as the reference group.
Figure 1. Kaplan-Meier plot of time to first AC-determined NMOSD attack during the RCP in AQP4-IgG seropositive patients:

AC adjudication committee; AQP4-IgG anti-aquaporin-4 immunoglobulin G; CI confidence interval; NMOSD neuromyelitis optica spectrum disorders; RCP randomised control period.
Across the RCP and OLP, the annualised AC-determined NMOSD attack rate was analysed as a secondary endpoint and in AQP4-IgG seropositive patients treated with inebilizumab the result was 0.09.
Immunoglobulin G4-related disease (IgG4-RD)
The efficacy of inebilizumab for the treatment of IgG4-RD was studied in one randomised (1:1), double-blind, multicentre, 52-week placebo-controlled clinical trial that enrolled 135 adult patients with active IgG4-RD. Patients had active disease, defined by clinical, imaging, laboratory or biopsy features, and required treatment in the judgement of the physician. Eligible patients had newly diagnosed or recurrent IgG4-RD that required glucocorticoid (GC) treatment at screening, had a confirmed history of organ involvement at any time in the course of the disease, and met the 2019 ACR/EULAR classification criteria.
All potential flares during the study were assessed by the investigator and subsequently reviewed by a blinded, independent, adjudication committee, who determined whether the flare met one or more of the protocol-defined, organ-specific flare diagnostic criteria. Disease flare was defined as new/worsening signs or symptoms that were positively adjudicated and warranted treatment by the investigator. Absence of alternative diagnoses was required.
Patients received 300 mg IV of inebilizumab or placebo at Days 1, 15 and 183 of the RCP. Patients were at a uniform dose of glucocorticoids (GCs) at the time of randomisation (equivalent to 20 mg prednisone per day) and then began a prespecified taper of 5 mg per day every 2 weeks to discontinuation at the end of 8 weeks. The use of GCs during the trial was permitted for treatment of IgG4-RD flares, and for other purposes including premedication for investigational treatment, oral GC treatment up to 2 weeks, or at a dose of up to 2.5 mg per day of prednisone or equivalent for treatment of adrenal insufficiency. The concomitant use of biologic and non-biologic immunosuppressive agents was prohibited during the trial. Patients who completed the RCP had the option to enrol in an OLP and initiate or continue treatment with inebilizumab.
227 patients were screened for eligibility. Of the 135 enrolled IgG4-RD patients, 68 patients were randomised to receive inebilizumab and 67 were randomised to receive placebo. Baseline demographics and disease characteristics for IgG4-RD patients during the RCP were balanced across the treatment groups (see table 6). Although no comparator was available during the OLP, treated and AC-determined flares in the open-label treatment period were determined.
Table 6. Demographics and baseline characteristics of IgG4-RD patients:
| Characteristic | Placebo N=67 | Inebilizumab N=68 | Overall N=135 |
|---|---|---|---|
| Age (years): mean (standard deviation [SD]) | 58.2 (12.2) | 58.2 (11.5) | 58.2 (11.8) |
| Age ≥65 years, n (%) | 21 (31.3%) | 21 (30.9%) | 42 (31.1%) |
| Sex: Male, n (%) | 49 (73.1%) | 39 (57.4%) | 88 (65.2%) |
| Disease duration (years): mean (SD) | 2.54 (3.06) | 2.64 (3.73) | 2.59 (3.40) |
| Ig-G4 manifestation Newly diagnosed | 31 (46.3%) | 31 (45.6%) | 62 (45.9%) |
| ACR/EULAR Classification Criteria score Mean (SD) | 38.3 (11.7) | 40.1 (12.1) | 39.2 (11.9) |
| Prior non-glucocorticoid therapy for IgG4-RD Yes | 20 (29.9%) | 17 (25.0%) | 37 (27.4%) |
| Baseline IgG4-RD Responder Index score Mean (SD) | 6.0 (4.0) | 5.4 (4.0) | 5.7 (4.0) |
Results in IgG4-RD patients are presented in figure 2 and table 7.
The study met the primary efficacy endpoint, time to the first treated and AC-determined IgG4-RD flare, which was longer in the inebilizumab group, compared with the placebo group (hazard ratio: 0.13; p<0.0001; see figure 2). The key secondary endpoints were also met with statistical significance (see table 7).
Figure 2. Primary endpoint - Kaplan-Meier plot of time to first treated and AC-determined IgG4-RD flare during the randomised-controlled period:
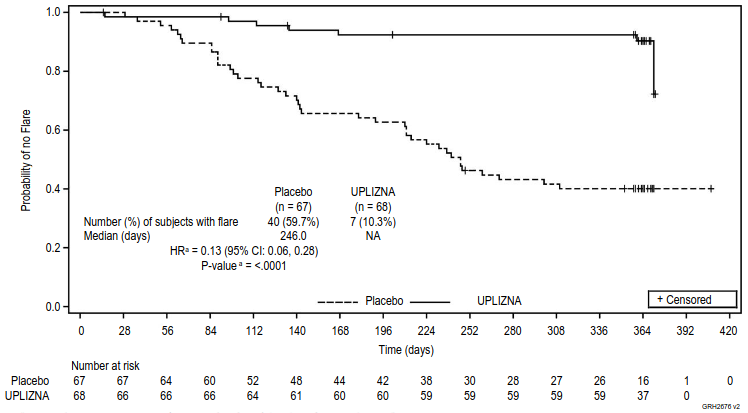
a Based on Cox regression method, with placebo as the reference group.
Patients who did not complete the RCP and who did not have a treated and AC-determined flare during the RCP were censored at the time of discontinuation.
Table 7. Key secondary efficacy results in IgG4-RD patients:
| Treatment group | ||
|---|---|---|
| Inebilizumab N=68 | Placebo N=67 | |
| Annualised flare rate for treated and AC-determined IgG4-RD flares | 0.10 | 0.71 |
| Rate ratio (95% CI)a | 0.14 (0.06, 0.31) | |
| p-valuea | <0.0001 | |
| Proportion of subjects achieving treatment-free, flare-free complete remission at week 52b | 39 (57.4%) | 15 (22.4%) |
| Odds ratio (95% CI)c | 4.68 (2.21, 9.91) | |
| p-valuec | <0.0001 | |
| Proportion of subjects achieving corticosteroid-free, flare-free complete remission at week 52d | 40 (58.8%) | 15 (22.4%) |
| Odds ratio (95% CI)c | 4.96 (2.34, 10.52) | |
| p-valuec | <0.0001 | |
a Estimated from the negative binomial regression, with placebo as the reference group.
b Defined as the lack of evident disease activity (IgG4-RD RI = 0 or investigator's decision) at week 52, no AC-determined flare during the RCP, and no treatment for flare or disease control except the required 8-week GC taper.
c Based on logistic regression model, with placebo as the reference group.
d Defined as the lack of evident disease activity (IgG4-RD RI = 0 or investigator's decision) at week 52, no AC-determined flare during the RCP, and no corticosteroid treatment for flare or disease control except the required 8-week GC taper.
The mean (SD) total GC use for IgG4-RD disease control per patient was lower in the inebilizumab group compared with the placebo group, with a mean (SD) of 118.25 (438.97) mg prednisone equivalent versus 1384.53 (1723.26) mg prednisone equivalent, respectively during the RCP. The mean (SD) daily GC use during the RCP per patient using GC was 3.34 (2.09) mg prednisone equivalent in the inebilizumab group versus 5.97 (4.20) mg prednisone equivalent in the placebo group. The mean (SD) total GC use during the RCP per patient using GC was 1148.71 (877.92) mg prednisone equivalent in the inebilizumab group versus 2208.65 (1707.56) mg prednisone equivalent in the placebo group.
Available data from the OLP, in which patients continued to receive inebilizumab supports a sustained treatment effect of inebilizumab.
Generalized myasthenia gravis (gMG)
The efficacy of inebilizumab for the treatment of gMG was studied in a randomised, double-blind, multicentre, placebo-controlled clinical trial. The RCP was 52 weeks for the anti-AChR-Ab+ population who received 300 mg IV of inebilizumab or placebo at Days 1, 15 and 183. The RCP was 26 weeks for the anti-MuSK-Ab+ population who received 300 mg IV of inebilizumab or placebo at Days 1 and 15. The primary analysis was conducted after week 26 in both populations.
Patients met the following eligibility criteria:
- Presence of autoantibodies against AChR or MuSK
- Myasthenia Gravis Foundation of America (MGFA) Clinical Classification Class II to IV
- Myasthenia Gravis-Activities of Daily Living (MG-ADL) score between 6 and 10 with >50% of this score attributed to non-ocular items or an MG-ADL score ≥11
- Quantitative Myasthenia Gravis (QMG) score of ≥11
- On a stable dose of a corticosteroid or a specified non-steroidal immunosuppressive therapy (NSIST), or a combination of both prior to randomisation.
The stable dose of corticosteroid (>5 mg/day prednisone or equivalent) was tapered to 5 mg/day (prednisone or equivalent) from week 4 to week 24. Rescue therapy included IVIg and plasma exchange.
Inebilizumab was administered according to the recommended dosage regimen (see section 4.2).
Patients were randomised at a 1:1 ratio; of the 238 enrolled gMG patients, 95 anti-AChR-Ab+ patients and 24 anti-MuSK-Ab+ patients were randomised to receive inebilizumab and 95 anti-AChR-Ab+ patients and 24 anti-MuSK-Ab+ patients were randomised to receive placebo.
Baseline demographics and disease characteristics for gMG patients during the RCP were balanced across the treatment groups (see Table 8).
Table 8. Demographics and baseline characteristics of gMG patients for overall population:
| Characteristic | Placebo N=117 | Inebilizumab N=119 | Overall N=236 |
| Age (years): mean (standard deviation [SD]) | 47.9 (15.0) | 47.1 (15.7) | 47.5 (15.3) |
| Age ≥65 years, n (%) | 16 (13.7) | 22 (18.5) | 38 (16.1) |
| Sex: Male, n (%) | 52 (44.4) | 40 (33.6) | 92 (39.0) |
| Sex: Female, n (%) | 65 (55.6) | 79 (66.4) | 144 (61.0) |
| Race, (%) | |||
| Asian | 46.2 | 38.7 | 42.4 |
| Black or African American | 2.6 | 1.7 | 2.1 |
| White | 47.9 | 58 | 53 |
| Disease duration (years): mean (SD) | 6.73 (7.28) | 5.94 (6.96) | 6.34 (7.12) |
| Baseline MG-ADL Score: mean (SD) | |||
| Overall population | 9.1 (2.8) | 9.0 (2.8) | 9.1 (2.8) |
| AChR+ population | 9.3 (2.8) | 9.1 (2.7) | 9.2 (2.7) |
| MuSK+ population | 8.3 (2.5) | 8.8 (3.1) | 8.5 (2.8) |
| Baseline QMG Score: mean (SD) | |||
| Overall population | 17.3 (4.2) | 16.7 (4.2) | 17.0 (4.2) |
| AChR+ population | 17.4 (4.3) | 16.9 (4.1) | 17.1 (4.2) |
| MuSK+ population | 17.2 (3.9) | 16.1 (4.8) | 16.7 (4.3) |
| Number of patients receiving stable immunosuppressant therapy at baseline: n (%) | |||
| Corticosteroid only | 68 (58.1) | 82 (68.9) | 150 (63.6) |
| NSIST only | 9 (7.7) | 8 (6.7) | 17 (7.2) |
| Corticosteroid plus 1 NSIST | 39 (33.3) | 29 (24.4) | 68 (28.8) |
| Acetylcholinesterase inhibitor: n (%) | 93 (79.5) | 94 (79.0) | 187 (79.2) |
The results of the primary and key secondary endpoints are presented in Table 9 and Figures 3 to 5.
The efficacy of inebilizumab was measured using the MG-ADL scale, which assesses the impact of gMG on an 8-item questionnaire that focuses on the MG patient's relevant symptoms and functional performance of activities of daily living. Each item is assessed on a 4-point scale where a score of 0 represents normal function and a score of 3 represents the most severe impact by the disease. The total MG-ADL score ranges from 0 to 24, with higher scores indicating more impairment.
The primary efficacy endpoint was the change from baseline in the MG-ADL score at week 26, in the overall population. A statistically significant difference favoring inebilizumab was observed in the mean change from baseline in MG-ADL total score (-4.3 inebilizumab vs -3.0 for placebo, a difference of -1.3, 95% CI: -2.2, -0.4; p-value: 0.0067). The proportion of patients who received rescue therapy by week 26 was lower in the inebilizumab group compared with placebo (8.4% inebilizumab vs 23.9% placebo).
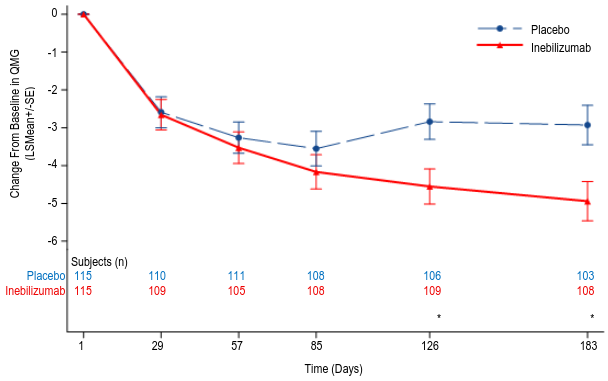
The key secondary endpoint was the change from baseline in the QMG score at week 26 in the overall population. The QMG score is a 13-item categorical grading system that quantitively measures disease impairment by mainly assessing muscle weakness. Each item is assessed on a 4-point scale where a score of 0 represents no impairment and a score of 3 represents severe impairment. A total possible score ranges from 0 to 39, where higher scores indicate more severe impairment. A statistically significant difference favoring inebilizumab was observed in the mean change from baseline in QMG total score (-4.9 inebilizumab vs -2.9 placebo, a difference of -2.0, 95% CI: -3.3, -0.7).
Table 9. Change from baseline in MG-ADL and QMG scores at week 26 in adult gMG patients who are Anti-AChR-Ab+ or Anti-MuSK-Ab+:
| Overall Population | ||
| Inebilizumab N=119 | Placebo N=117 | |
| MG-ADL Score | ||
| LS Mean | -4.3 | -3.0 |
| Difference | -1.3 | |
| 95% CI | (-2.2, -0.4) | |
| p-value | 0.0067 | |
| QMG Score | ||
| LS Mean | -4.9 | -2.9 |
| Difference | -2.0 | |
| 95% CI | (-3.3, -0.7) | |
| p-value | 0.0028 | |
CI = confidence interval; LS = least square
The proportion of patients who achieved a ≥3-point improvement in MG-ADL score at week 26 with no use of rescue therapy between Day 28 and week 26 was 68.7% in the inebilizumab group and 48.2% in the placebo group.
Figure 3. Mean change from baseline in MG-ADL score by week 26 for overall population:
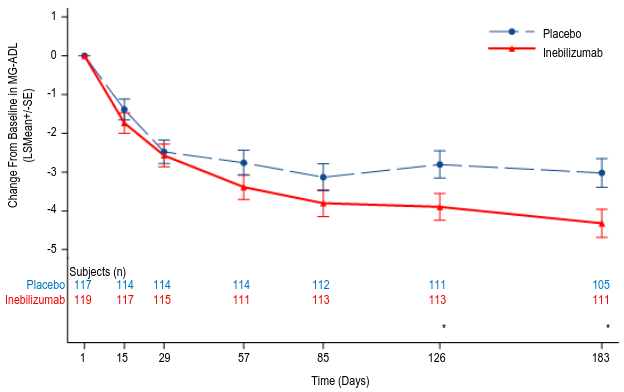
Figure 4. Mean change from baseline in QMG score by week 26 for overall population:
Patients who were gMG anti-AChR-Ab+ continued in the RCP until week 52. Results in these patients show that the treatment difference favoring inebilizumab increased over time, compared with placebo. At week 52, in the anti-AChR-Ab+ population, the change from baseline in the MG-ADL score was -5.1 vs -3.0 for inebilizumab and placebo, respectively.
Figure 5. Mean change from baseline in MG-ADL score by week 52 for anti-AChR-Ab+ patients:
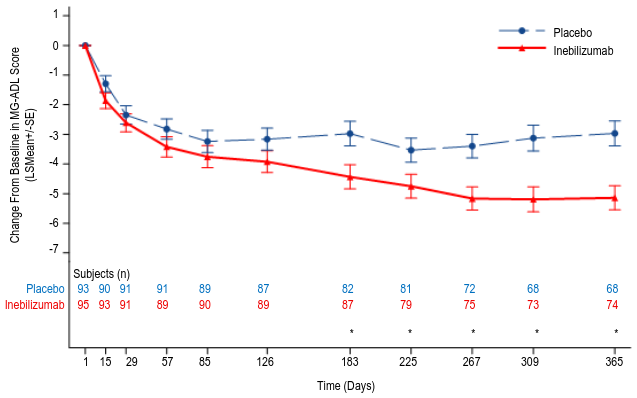
At the time of primary analysis, in total, 94 patients (79.0%) in the inebilizumab group and 84 patients (70.6%) in the placebo group received any dose of inebilizumab during the OLP. Among patients who initially received inebilizumab during the RCP, continued improvements in MG-ADL and QMG were observed through week 78 of OLP in the anti-AChR-Ab+ and in the anti-MuSK-Ab+ subpopulations, respectively. Among patients who initially received placebo and initiated treatment with inebilizumab during the OLP, improvements in MG-ADL and QMG continued through week 78 of OLP in the anti-AChR-Ab+ and in the anti-MuSK-Ab+ subpopulations, respectively.
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with inebilizumab in one or more subsets of the paediatric population in NMOSD, IgG4-RD and gMG (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Absorption
Inebilizumab is administered as an intravenous infusion. In the NMOSD study, the mean maximum concentration was 108 μg/mL (300 mg, second dose on day 15), and the cumulative area under the curve (AUC) of the 26-week treatment period in which NMOSD patients received two intravenous administrations 2-week apart was 2980 μg×d/mL. In the IgG4-RD study, the mean maximum concentration was 127 μg/mL (300 mg, second dose on day 15), and the cumulative AUC of the 52-week treatment period in which IgG4-RD patients received two intravenous administrations 2 weeks apart, followed by a third dose at week 26 was 4290 μg×d/mL.
In the gMG study, the mean maximum concentration (300 mg, second dose on day 15) was 126 μg/mL for the anti-AChR-Ab+ subpopulation (n=90) and 159 μg/mL for the anti-MuSK-Ab+ subpopulation (n=20). The cumulative AUC of the 26-week treatment period in which anti-AChR-Ab+ patients (n=82) received two intravenous administrations 2 weeks apart was 3120 μg×d/mL. The cumulative AUC of the 26-week treatment period in which anti-MuSK-Ab+ patients (n=17) received two intravenous administrations 2 weeks apart was 3740 μg×d/mL. Additionally, the cumulative AUC of the 52-week treatment period in which anti-AChR-Ab+ patients (n=84) received two intravenous administrations 2 weeks apart, followed by a third dose at week 26 was 4240 μg×d/mL.
Distribution
Based on population pharmacokinetic analysis, the estimated typical central and peripheral volume of distribution of inebilizumab was 2.95 L and 2.57 L, respectively.
Biotransformation
Inebilizumab is a humanised IgG1 monoclonal antibody that is degraded by proteolytic enzymes widely distributed in the body.
Elimination
In adult patients with NMOSD, IgG4-RD and gMG, the terminal elimination half-life was approximately 18 days. From population pharmacokinetic analysis, the estimated inebilizumab systemic clearance of the first-order elimination pathway was 0.19 L/day. At low pharmacokinetic exposure levels, inebilizumab was likely subject to the receptor (CD19)-mediated clearance, which decreased with time presumably due to the depletion of B cells by inebilizumab treatment.
Special populations
Paediatric population
Inebilizumab has not been studied in adolescents or children.
Elderly
Based on population pharmacokinetic analysis, age did not affect inebilizumab clearance.
Gender, race
A population pharmacokinetic analysis indicated that there was no significant effect of gender and race on inebilizumab clearance.
Renal impairment
No formal clinical studies have been conducted to investigate the effect of renal impairment on inebilizumab. Due to the large molecular weight and hydrodynamic size of an IgG monoclonal antibody, inebilizumab is not expected to be filtered through the glomerulus. From population pharmacokinetic analysis, inebilizumab clearance in patients with varying degrees of renal impairment was comparable to patients with normal estimated glomerular filtration rate.
Hepatic impairment
No formal clinical studies have been conducted to investigate the effect of hepatic impairment on inebilizumab. In clinical studies, no subjects with severe hepatic impairment have been exposed to inebilizumab. IgG monoclonal antibodies are not primarily cleared via the hepatic pathway; change in hepatic function is, therefore, not expected to influence inebilizumab clearance. Based on population pharmacokinetic analysis, baseline hepatic function biomarkers (AST, ALP, and bilirubin) had no clinically relevant effect on inebilizumab clearance.
5.3. Preclinical safety data
Nonclinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, and carcinogenic potential.
Inebilizumab was evaluated in a combined fertility and embryo-foetal development study in female and male huCD19 Tg mice at intravenous doses of 3 and 30 mg/kg. There was no effect on embryo-foetal development, however, there was a treatment-related reduction in fertility index at both tested doses. The relevance of this finding to humans is unknown. Additionally, there was a decrease in B-cell populations at the site of B-cell development in foetal mice born to inebilizumab-treated animals as compared to the offspring of control animals, suggesting that inebilizumab crosses the placenta and depletes B cells.
Only sparse toxicokinetic samples were collected in the combined fertility and embryo-foetal development study; based on first dose maximum concentration (Cmax), the exposure multiples of 3 and 30 mg/kg in female huCD19 Tg mice were 0.4-fold and 4-fold, respectively for the 300 mg clinical therapeutic dose.
In a pre-/postnatal development study in transgenic mice, administration of inebilizumab to maternal animals from gestation day 6 to lactation day 20 resulted in depleted B-cell populations in offspring at postnatal day 50. B-cell populations in offspring recovered by postnatal day 357. The immune response to neoantigen in offspring of animals treated with inebilizumab was decreased relative to offspring of control animals, suggestive of impairment of normal B-cell function.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.