Source: European Medicines Agency (EU) Revision Year: 2019 Publisher: Vifor Fresenius Medical Care Renal Pharma France, 100–101 Terrasse Boieldieu, Tour Franklin La Défense 8, 92042 Paris La Défense Cedex, France
Pharmacotherapeutic group: Drugs for treatment of hyperkalaemia and hyperphosphataemia
ATC code: V03AE09
Patiromer is a non-absorbed, cation exchange polymer that contains a calcium-sorbitol complex as a counterion.
Patiromer increases faecal potassium excretion through binding of potassium in the lumen of the gastrointestinal tract. Binding of potassium reduces the concentration of free potassium in the gastrointestinal lumen, resulting in a reduction of serum potassium levels.
In healthy adult subjects, patiromer caused a dose dependent increase in faecal potassium excretion, and a corresponding decrease in urinary potassium excretion with no change in serum potassium. 25.2 g of patiromer, administered once daily for 6 days, resulted in a mean increase in faecal potassium excretion of 1283 mg/day, and a mean decrease in urinary potassium excretion of 1438 mg/day. Daily urinary calcium excretion increased from baseline by 53 mg/day.
In an open label study to assess the time to onset of action, a statistically significant reduction in serum potassium in hyperkalaemic patients was observed at 7 hours after the first dose. Following discontinuation of patiromer, potassium levels remained stable for 24 hours after the last dose, then rose again during a 4-day observation period.
The safety and efficacy of patiromer were demonstrated in a two-part, single blind randomised withdrawal study that evaluated this treatment in hyperkalaemic patients with chronic kidney disease (CKD) on stable doses of at least one RAAS inhibitor (i.e. angiotensin converting enzyme inhibitor [ACEI], angiotensin II receptor blocker [ARB] or aldosterone antagonist [AA]).
In Part A, 243 patients were treated with patiromer for 4 weeks. Patients with a baseline serum potassium of 5.1 mEq/L to <5.5 mEq/L (mmol/L) received a starting dose of 8.4 g patiromer per day (as a divided dose) and patients with a baseline serum potassium of 5.5 mEq/L to <6.5 mEq/L received a starting dose of 16.8 g patiromer per day (as a divided dose). The dose was titrated, as needed, based on the serum potassium level, assessed starting on Day 3 and then at weekly visits to the end of the 4 week treatment period, with the aim of maintaining serum potassium in the target range (3.8 mEq/L to <5.1 mEq/L). The mean daily doses of patiromer were 13 g and 21 g in patients with serum potassium of 5.1 to <5.5 mEq/L and 5.5 to <6.5 mEq/L, respectively.
The mean age of patients was 64 years (54% aged 65 and over, 17% aged 75 and over), 58% of patients were men, and 98% were Caucasian. Approximately 97% of patients had hypertension, 57% had type 2 diabetes, and 42% had heart failure.
Mean serum potassium levels and change in serum potassium from Part A Baseline to Part A Week 4 is shown in Table 1. For the Part A secondary outcome, 76% (95% CI: 70%, 81%) of patients had a serum potassium in the target range of 3.8 mEq/L to <5.1 mEq/L at Part A Week 4.
Table 1. Patiromer treatment phase (Part A): Primary endpoint:
| Baseline Potassium | Overall Population (n=237) | ||
|---|---|---|---|
| 5,1 έως <5,5 mEq/l | 5,5 έως <6,5 mEq/l (n=147) | ||
| Serum Potassium (mEq/l) | |||
| Baseline, mean (SD) | 5.31 (0.57) | 5.74 (0.40) | 5.58 (0.51) |
| Week 4 Change from Baseline, Mean ± SE (95% CI) | −0.65 ± 0.05 | −1.23 ± 0.04 | −1.01 ± 0.03 |
| (−0.74, −0.55) | (−1.31, −1.16) | (−1.07, −0.95) | |
| p value | <0.001 | ||
In Part B, 107 patients with a Part A baseline serum potassium of 5.5 mEq/L to <6.5 mEq/L and whose serum potassium was in the target range (3.8 mEq/L to <5.1 mEq/L) at Part A Week 4 and still receiving RAAS inhibitor treatment were randomised to continue patiromer or to receive placebo for 8 weeks to evaluate the effect of withdrawing patiromer on serum potassium. In patients randomised to patiromer, the mean daily dose was 21 g at the start of Part B and during Part B.
The Part B primary endpoint was the change in serum potassium from Part B baseline to the earliest visit at which the patient’s serum potassium was first outside of the range of 3.8 to <5.5 mEq/L or to Part B Week 4 if the patient’s serum potassium remained in the range. In Part B, serum potassium in patients on placebo increased significantly relative to patients who remained on patiromer (p<0.001).
More placebo patients (91% [95% CI: 83%, 99%]) developed a serum potassium ≥5.1 mEq/L at any time during Part B than patiromer patients (43% [95% CI: 30%, 56%]), p<0.001. More placebo patients (60% [95% CI: 47%, 74%]) developed a serum potassium ≥5.5 mEq/L at any time during Part B than patiromer patients (15% [95% CI: 6%, 24%]), p<0.001.
The potential of patiromer to enable concomitant RAAS inhibitor treatment was also assessed in part B. Fifty two percent (52%) of subjects receiving placebo discontinued RAAS inhibitor treatment because of recurrent hyperkalaemia compared with 5% of subjects treated with patiromer.
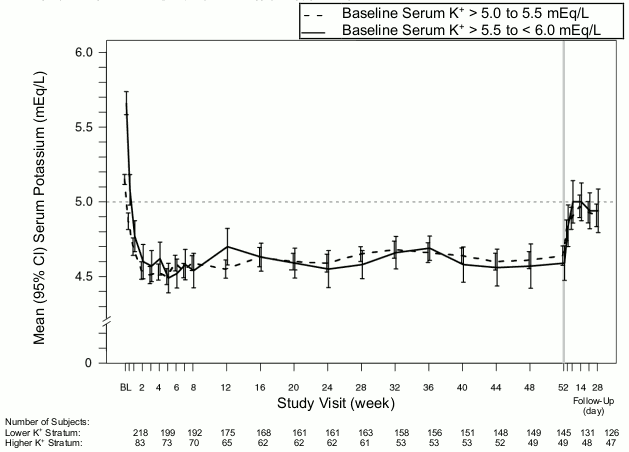
The effect of treatment with patiromer for up to 52 weeks was evaluated in an open label study of 304 hyperkalaemic patients with CKD and type 2 diabetes mellitus on stable doses of a RAAS inhibitor. The mean age of patients was 66 years (59.9% aged 65 and over, 19.7% aged 75 and over), 63% of patients were men, and all were Caucasian. Decreases in serum potassium with patiromer treatment were maintained over 1 year of chronic treatment as shown in Figure 1, with a low incidence of hypokalaemia (2.3%) and the majority of subjects reaching (97.7%) and maintaining target serum potassium levels (overall during maintenance period, serum potassium was within the target range for approximately 80% of the time). In patients with a baseline serum potassium of >5.0 to 5.5 mEq/L who received an initial dose of 8.4 g patiromer per day, the mean daily dose was 14 g; in those with a baseline serum potassium of >5.5 to <6.0 mEq/L who received an initial dose of 16.8 g patiromer per day, the mean daily dose was 20 g during the entire study.
Figure 1. Mean (95% CI) serum potassium over time:
The ability of patiromer to enable concomitant spironolactone treatment was investigated in a randomised, double-blind, placebo-controlled study in heart failure patients who were clinically indicated to receive AA. Patients initiated spironolactone at 25 mg/day at the same time as their randomised treatment (patiromer 12.6 g BID or placebo), and were up-titrated to 50 mg/day after Day 14 if serum potassium was >3.5 and ≤5.1 mEq/L. Of the 105 patients who were randomised and received study treatment (patiromer 56; placebo 49), mean age was 68.3 years, 60.6% were men, 97.1% were Caucasian, and mean eGFR was 81.3 mL/min. Mean baseline serum potassium values were 4.71 mEq/L for patiromer and 4.68 mEq/L for placebo. The primary efficacy endpoint, change from baseline in serum potassium to the end of the 28-day treatment period, was significantly lower (p<0.001) in the patiromer group (LS mean [SEM]: −0. 21 [0.07] mEq/L) as compared to the placebo group (LS mean [SEM]: +0.23 [0.07] mEq/L). There were also fewer patients in the patiromer group with serum potassium values >5.5 mEq/L (7.3% vs. 24.5%; p=0.027) and more patients on spironolactone 50 mg/day (90.9% versus 73.5%, p=0.022).
Overall, in the phase 2 and 3 clinical studies, 99.4% of patients were receiving RAAS inhibitor therapy at baseline, 81.2% had CKD with eGFR <60 mL/min/1.73 m², 72.8% had diabetes mellitus and 48.7% had heart failure.
The European Medicines Agency has deferred the obligation to submit the results of studies with patiromer in one or more subsets of the paediatric population in the treatment of hyperkalaemia (see section 4.2 for information on paediatric use).
In an open-label study, 114 patients with hyperkalaemia were randomized to patiromer once daily with food or without food. Serum potassium at the end of treatment, the change from baseline in serum potassium, and the mean dose of patiromer were similar between groups.
Patiromer works by binding potassium in the gastrointestinal tract and thus the serum concentration is not relevant for its efficacy. Due to the insolubility and nonabsorptive characteristics of this medicinal product, many classical pharmacokinetic studies cannot be carried out.
Patiromer is excreted approximately 24 to 48 hours after intake, based on average gastrointestinal transit time.
In radiolabeled studies in rats and dogs, patiromer was not systemically absorbed and was excreted in the faeces. Quantitative whole-body autoradiography analysis in rats demonstrated that radioactivity was limited to the gastrointestinal tract, with no detectable level of radioactivity in any other tissues or organs.
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, toxicity to reproduction and development.
Patiromer was not genotoxic in the reverse mutation test (Ames assay), chromosome aberration or rat micronucleus assays.
Carcinogenicity studies have not been performed.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.