VEPPANU Film-coated tablet Ref.[116672] Active ingredients: Vepdegestrant
Source: FDA, National Drug Code (US) Revision Year: 2025
12.1. Mechanism of Action
Vepdegestrant is a heterobifunctional protein degrader that binds to estrogen receptor (ER) and the E3 ligase cereblon (CRBN). This interaction results in the degradation cascade through CRBN-mediated polyubiquitination and degradation of ER by the proteasome, leading to reduction of ER protein levels in breast cancer cells.
Vepdegestrant induced degradation of wild-type (WT) and mutant ER, inhibited ER-dependent breast cancer cell line proliferation in vitro and demonstrated antitumor activity in vivo in both WT and mutant ESR1 breast cancer models.
12.2. Pharmacodynamics
The exposure-response relationship and time-course of pharmacodynamic response of vepdegestrant have not been fully characterized.
Cardiac Electrophysiology
The largest mean increase in QTc interval was 12 ms (upper confidence interval = 15 ms) after administration of vepdegestrant at the recommended dosage of 200 mg once daily in patients with ESR1 mutation-positive breast cancer.
12.3. Pharmacokinetics
Vepdegestrant pharmacokinetics were observed at steady state in patients with ER+/HER2- breast cancer at the approved recommended dosage of 200 mg once daily and are presented as mean (coefficient of variation (CV%)) unless otherwise specified.
Vepdegestrant maximum concentration (Cmax) is 926 ng/mL (39%), and the total systemic exposure (AUC) is 17155 ng•hr/mL (36%). Vepdegestrant AUC and Cmax increase in an approximately dose proportional manner over the dose range of 100 mg (0.5 times the approved recommended dose) to 500 mg (2.5 times the approved recommended dose). Vepdegestrant accumulation is approximately 1.4-fold for AUC and 1.3-fold for Cmax. Vepdegestrant steady state is reached in approximately 7 days.
Absorption
Vepdegestrant median (min, max) time to maximum plasma concentration (Tmax) is approximately 6 (4, 8) hours.
Effect of Food
Vepdegestrant AUC increased 2.9-fold and Cmax 3.2-fold following administration with a high-fat meal (approximately 800 to 1,000 calories; ≥50% fat).
Distribution
Vepdegestrant plasma protein binding is >99%. The apparent (oral) volume of distribution is 764 L (26%) following a single 200 mg dose.
Elimination
Vepdegestrant effective elimination half-life is 19 hours (50%) with an apparent (oral) clearance of 12 L/h (36%).
Metabolism
Vepdegestrant is primarily metabolized through direct sulfation via multiple SULT isoforms and oxidation via CYP3A4 and to a lesser extent by CYP2C8, CYP2C9 and CYP3A5. Unchanged total vepdegestrant represented 92% of total radioactivity in plasma.
Excretion
Following a single oral dose of radiolabeled vepdegestrant 200 mg to healthy subjects, approximately 68% of the dose was recovered in feces (18% unchanged) and 1.5% in urine (<0.02% unchanged).
Specific Populations
No clinically significant differences in the pharmacokinetics of vepdegestrant were observed based on age (26 to 89 years), sex, body weight (37 to 181 kg), race (60% White, 27% Asian, 2.2% Black or African American, and 10% other), CLcr 30 mL/min to 90 mL/min (estimated by Cockcroft Gault equation) or mild hepatic impairment (total bilirubin ≤ULN and AST>ULN or total bilirubin >1 to 1.5 × ULN and any AST).
The effect of moderate hepatic impairment (total bilirubin >1.5 to 3 × ULN and any AST), severe hepatic impairment (total bilirubin >3 x ULN and any AST), CLcr 15 to <30 mL/min, and end-stage renal disease (CLcr <15 mL/min) on the pharmacokinetics of vepdegestrant is unknown.
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
Strong CYP3A Inhibitors: Vepdegestrant AUC increased 1.7-fold and Cmax 1.5-fold following concomitant use of itraconazole (strong CYP3A inhibitor) 200 mg once daily.
Strong CYP3A Inducers: Vepdegestrant AUC decreased to 64% and Cmax to 80% following concomitant use of carbamazepine (strong CYP3A inducer) 200 mg three times daily.
Acid Reducing Agents: Vepdegestrant AUC decreased to 84% and Cmax to 74% following concomitant use of esomeprazole (proton-pump inhibitor) 40 mg once daily.
CYP3A Substrates: Midazolam (CYP3A substrate) AUC increased 1.7-fold and Cmax 1.2-fold following concomitant use of VEPPANU 200 mg once daily.
P-gp Substrates: Dabigatran etexilate (P-gp substrate) AUC increased 2-fold and Cmax 1.9-fold following concomitant use of a single dose of VEPPANU 200 mg.
Breast Cancer Resistance Protein (BCRP) Substrates: Rosuvastatin (BCRP substrate) AUC increased 1.2-fold and Cmax 1.2-fold following concomitant use of a single dose of VEPPANU 200 mg.
UGT1A9 Substrates: Dapagliflozin (UGT1A9 index substrate) AUC is predicted to increase approximately 2‑fold following multiple doses of VEPPANU.
In vitro studies
CYP450 Enzymes: Vepdegestrant does not inhibit CYP1A2, CYP2C8, CYP2C9, CYP2C19 or CYP2D6 and does not induce CYP1A2, CYP2C8, CYP2C9, or CYP2C19.Vepdegestrant is an inhibitor of CYP2B6.
UDP-Glucuronosyltransferases (UGT): Vepdegestrant does not inhibit UGT1A1, UGT1A4, UGT1A6, UGT2B7, or UGT2B15.
Transporter Systems: Vepdegestrant is not a substrate of P-gp, BCRP, OATP1B1, or OATP1B3.
Vepdegestrant does not inhibit MATE1, MATE2-K, OAT1, OAT3, OATP1B1, OATP1B3, or OCT2.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Vepdegestrant was not carcinogenic in a 6-month carcinogenicity study in rasH2 transgenic mice with daily oral administration of vepdegestrant up to 800 mg/kg/day.
Mutagenesis
Vepdegestrant was not mutagenic in an in vitro bacterial reverse mutation assay (Ames) or clastogenic in an in vitro human lymphocyte micronucleus assay or an in vivo rat micronucleus assay.
Impairment of Fertility
Fertility studies with vepdegestrant in animals have not been conducted. In repeat-dose toxicity studies up to 26 weeks duration in rats and 39 weeks duration in dogs, oral administration of vepdegestrant resulted in adverse female reproductive effects including atrophy of the uterus, oviduct, cervix and vagina and follicular cysts in rats at doses ≥30 mg/kg/day (0.3 times the human AUC at the recommended dose) and in dogs at doses ≥10 mg/kg/day (0.4 times the human AUC at the recommended dose). Additional findings in the ovary in rats included decreased corpora lutea and follicle hemorrhage at ≥30 mg/kg/day. Oral administration of vepdegestrant resulted in adverse male reproductive effects including decreased epididymis and prostate weights with decreased secretion and decreased secretion in the seminal vesicle in rats at doses ≥30 mg/kg/day, and seminiferous tubular degeneration, hypoplasia of seminiferous tubules, and epididymal epithelial cell necrosis in dogs at 90 mg/kg/day (approximately 2 times the human AUC at the recommended dose). The effects of vepdegestrant on female reproductive organs were reversible following a 4-week recovery period. The effects on male reproductive organs were not reversible following a 4-week recovery period.
13.2. Animal Toxicology and/or Pharmacology
In a 6-month repeat-dose toxicity study, oral administration of vepdegestrant to rats resulted in granulosa cell hyperplasia in the ovary at 300 mg/kg/day (5 times the human AUC at the recommended dose). In a 9-month repeat-dose toxicity study, oral administration of vepdegestrant to dogs resulted in interstitial cell hypertrophy/hyperplasia in the testis at doses ≥10 mg/kg/day (0.4 times the human AUC at the recommended dose). Reversibility was not assessed.
14. Clinical Studies
The efficacy of VEPPANU was evaluated in VERITAC-2 (NCT05654623), a randomized, open-label, active-controlled, multicenter trial that enrolled 624 adult patients with ER-positive, HER2-negative, advanced or metastatic breast cancer, of whom 270 patients had tumors carrying ESR1 mutations. Patients were required to have disease progression on 1 to 2 prior lines of endocrine therapy, including 1 line with a CDK4/6 inhibitor. Progression during or within 12 months from the end of adjuvant therapy was counted as 1 line of endocrine therapy for advanced/metastatic setting. Pre-menopausal and peri-menopausal women and men received a gonadotropin-releasing hormone (GnRH) agonist. Patients were excluded if they had received chemotherapy for advanced or metastatic disease or fulvestrant in any line of therapy, or if progression on the most recent line of endocrine therapy occurred within the first 6 months.
Patients were randomized 1:1 to receive VEPPANU 200 mg orally once daily (N=313), or fulvestrant 500 mg intramuscularly on Days 1 and 15 of Cycle 1 and then once monthly thereafter (N=311). Randomization was stratified by ESR1 mutation status (detected vs. not detected) and visceral metastasis (yes vs. no). ESR1 mutational status was determined by blood circulating tumor deoxyribonucleic acid (ctDNA) using central or local testing. Patients were treated until disease progression or unacceptable toxicity.
Among the patients whose tumors had ESR1 mutations (N=270), the median age was 60 (range: 26 to 87) years; all but 1 were female; 47% were White; 41% Asian; 3.3% Black; 8% unknown/not reported; 7% were Hispanic/Latino, 83% were Not Hispanic or Latino and 10% not reported. Of the 269 women, 20% were pre/perimenopausal. Baseline Eastern Cooperative Oncology Group (ECOG) performance status was 0 (57%) or 1 (43%). Most patients (68%) had visceral disease; 81% had received 1 line of endocrine therapy and 19% had received 2 lines of endocrine therapy in the advanced or metastatic setting. All patients had received prior treatment with a CDK4/6 inhibitor.
The major efficacy outcome was progression-free survival (PFS) as assessed by blinded independent central review (BICR) in the population of patients whose tumors had an ESR1 mutation and in the overall population evaluated according to Response Evaluation Criteria in Solid Tumors (RECIST), version 1.1. Additional efficacy outcomes were overall survival (OS) and objective response rate (ORR) as assessed by BICR.
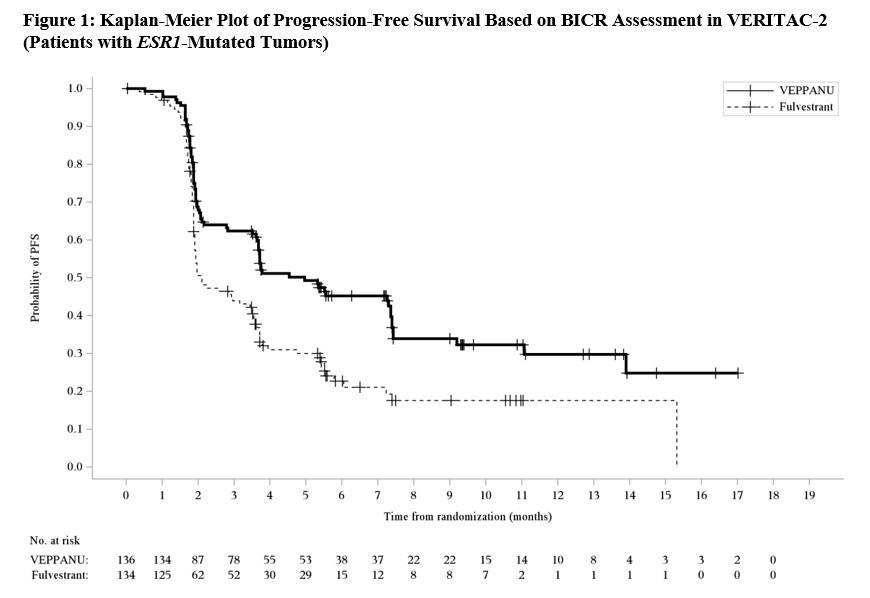
A statistically significant difference in PFS by BICR was observed for the patients whose tumors had ESR1 mutations for VEPPANU compared with fulvestrant.
Overall survival was immature with 16% of deaths in this population at the time of the PFS analysis. Efficacy results are provided in Table 7 and Figure 1.
Table 7. Efficacy Results for VERITAC-2 (Patients with ESR1-Mutated Tumors):
| VEPPANU N=136 | Fulvestrant N=134 | |
| Progression-free Survival* | ||
| Number of events (%) | 79 (58) | 95 (71) |
| Median in months (95% CI) | 5.0 (3.7, 7.4) | 2.1 (1.9, 3.5) |
| Hazard ratio (95% CI)a | 0.57 (0.42, 0.77) | |
| p-value (1-sided)b | 0.0001 | |
| Confirmed Objective Response Rate* | ||
| Patients with measurable disease | 97 | 100 |
| ORR (95% CI) | 19% (12, 27) | 4% (1.6, 10) |
| Complete response rate | 0% | 0% |
| Partial response rate | 19% | 4% |
Abbreviations: CI=Confidence interval; n=number of events; N=number of participants
* By blinded independent central review (BICR).
a Hazard ratio based on stratified Cox proportional hazards model.
b p-value based on stratified log-rank test (compared to a significance level of 0.01875).
Figure 1. Kaplan-Meier Plot of Progression-Free Survival Based on BICR Assement in VERITAC-2 (Patients with ESMR1-Mutated Tumors):
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.