Source: FDA, National Drug Code (US) Revision Year: 2020
The mechanism of action of vilazodone in the treatment of major depressive disorder is not fully understood, but is thought to be related to its enhancement of serotonergic activity in the CNS through selective inhibition of serotonin reuptake. Vilazodone is also a partial agonist at serotonergic 5-HT1A receptors; however, the net result of this action on serotonergic transmission and its role in vilazodone’s antidepressant effect are unknown.
Vilazodone binds with high affinity to the serotonin reuptake site (Ki= 0.1 nM), but not to the norepinephrine (Ki=56 nM) or dopamine (Ki=37 nM) reuptake sites. Vilazodone potently and selectively inhibits reuptake of serotonin (IC50= 1.6 nM). Vilazodone also binds selectively with high affinity to 5-HT1A receptors (IC50=2.1 nM) and is a 5-HT1A receptor partial agonist.
Treatment with VIIBRYD did not prolong the QTc interval. The effect of VIIBRYD [20, 40, 60, and 80 mg (2 times the recommended dosage)] on the QTc interval was evaluated in a randomized, placebo-, and active-controlled (moxifloxacin 400 mg), parallel-group, thorough QTc study in 157 healthy subjects. The study demonstrated an ability to detect small effects. The upper bound of the 90% confidence interval for the largest placebo-adjusted, baseline-corrected QTc interval was below 10 msec, based on the individual correction method (QTcI). Thus, at doses of 2 times the recommended dosage, VIIBRYD did not prolong the QTc interval to a clinically relevant extent.
Vilazodone activity is due primarily to the parent drug. The pharmacokinetics of vilazodone (5 mg – 80 mg) are dose-proportional. Accumulation of vilazodone after administration of single VIIBRYD doses did not vary with dose, and steady-state was achieved in about 3 days. Elimination of vilazodone is primarily by hepatic metabolism with a terminal half-life of approximately 25 hours. At steady-state, after daily dosing of VIIBRYD 40 mg under fed conditions, the mean Cmax value was 156 ng/mL, and the mean AUC(0-24 hours) value was 1645 ng·h/mL.
Vilazodone concentrations peaked at a median of 4-5 hours (Tmax) after VIIBRYD administration and declined with a terminal half-life of approximately 25 hours. The absolute bioavailability of vilazodone was 72% with food.Vilazodone AUC and Cmax in the fasted state can be decreased by approximately 50% and 60%, respectively, compared to the fed state. Administration without food can result in inadequate drug concentrations and may reduce effectiveness.
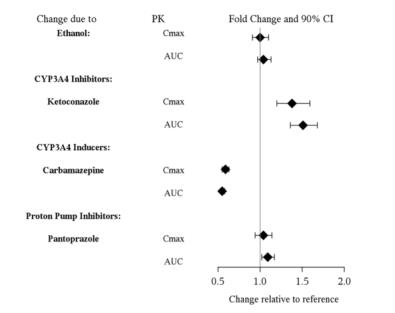
Coadministration of VIIBRYD with ethanol or with a proton pump inhibitor (pantoprazole) did not affect the rate or extent of vilazodone absorption. In addition, neither the Tmax nor terminal elimination rate of vilazodone was altered by coadministration with either pantoprazole or ethanol.
Absorption is decreased by approximately 25% if vomiting occurs within 7 hours of ingestion; no replacement dose is needed.
Vilazodone is widely distributed and approximately 96-99% protein-bound. Administration of VIIBRYD to a patient taking another drug that is highly protein bound may cause increased free concentrations of the other drug, because vilazodone is highly bound to plasma protein. The interaction between vilazodone and other highly protein-bound drugs has not been evaluated.
VIIBRYD is extensively metabolized through CYP and non-CYP pathways (possibly by carboxylesterase), with only 1% of the dose recovered in the urine and 2% of the dose recovered in the feces as unchanged vilazodone. CYP3A4 is primarily responsible for its metabolism among CYP pathways, with minor contributions from CYP2C19 and CYP2D6.
Figure 1 below includes the impact of other drugs on the pharmacokinetics of vilazodone [see Drug Interactions (7)].
Figure 1. Effect of Other Drugs on Vilazodone Pharmacokinetics:
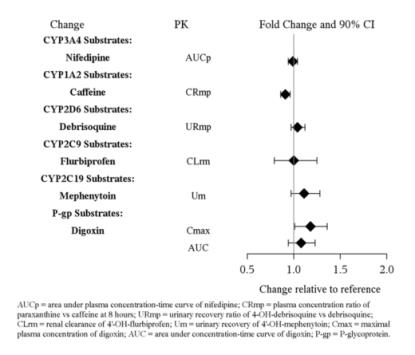
In vitro studies indicate that vilazodone is unlikely to inhibit or induce the metabolism of substrates for CYP1A1, 1A2, 2A6, 2B6, 2C9, 2C19, 2D6, 2E1, 3A4 or 3A5, except for CYP2C8. The effect of vilazodone on CYP2C8 activity has not been tested in vivo. Figure 2 below includes the impact of vilazadone on the pharmacokinetics of other drugs in vivo.
Figure 2. Impact of Vilazodone on Other Drug Pharmacokinetics:
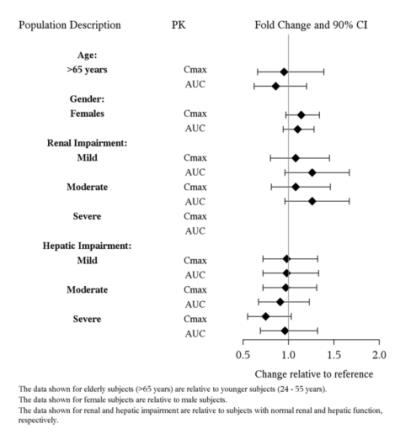
The presence of mild to severe renal impairment or mild to severe hepatic impairment did not affect the apparent clearance of vilazodone (see Figure 3). There were no pharmacokinetic differences of vilazodone in geriatric patients compared to younger patients, or between males and females (see Figure 3).
Figure 3. Impact of Intrinsic Factors on Vilazodone Pharmacokinetics:
Carcinogenicity studies were conducted in which B6C3F1mice and Wistar rats were given oral doses of vilazodone up to 135 and 150 mg/kg/day, respectively, for 2 years. These doses are approximately 16.5 and 36 times the maximum recommended human dose (MRHD) of 40 mg, respectively, on a mg/m2 basis.
In mice, the incidence of hepatocellular carcinomas was increased in males at 16.5 times the MRHD; this finding was not observed at 5.5 times the MRHD. The incidence of malignant mammary gland tumors was numerically increased in females at 5.5 and 16.5 times the MRHD, with statistical significance at 16.5 the MRHD; this finding was not observed at 1.8 times the MRHD. Elevated prolactin levels were observed in a 2-week study of vilazodone administered at 5.5 and 33 times the MRHD. Increases in prolactin levels are known to cause mammary tumors in rodents.
In the rat study, vilazodone was not carcinogenic in either sex at doses up to 36 times the MRHD.
Vilazodone was not mutagenic in the in vitro bacterial reverse mutation assay (Ames test). Vilazodone was negative in the in vitro V79/HGRPT mammalian cell forward mutation assay. Vilazodone was clastogenic in two in vitro mammalian cell chromosome aberration assays. However, vilazodone was negative for clastogenic activity in both an in vivo rat bone marrow chromosome aberration assay and a micronucleus test. Vilazodone was also negative in an in vivo/in vitro unscheduled DNA synthesis assay in rats.
Treatment of rats with vilazodone at a dose of 125 mg/kg, which is 30 times the MRHD of 40 mg on a mg/m2 basis, caused impairment of male fertility with no effect on female fertility. Impaired male fertility was not observed at 6 times the MRHD.
The efficacy of VIIBRYD as a treatment for major depressive disorder was demonstrated in four multicenter, randomized, double-blind, placebo-controlled studies in adult (18-70 years of age) outpatients who met the Diagnostic and Statistical Manual of Mental Disorders (DSM-IV-TR) criteria for MDD. Three 8-week studies evaluated the efficacy of VIIBRYD 40 mg (Studies 1-3) and one 10-week study (Study 4) evaluated the efficacy of VIIBRYD 20 mg and 40 mg (see Table 5). In these studies, patients were randomized to either 20 mg or 40 mg, or placebo once daily with food. Patients were either titrated over 1week to a dose of 20 mg daily or over 2 weeks to a dose of 40 mg once daily of VIIBRYD with food. VIIBRYD was superior to placebo in the improvement of depressive symptoms as measured by the change from baseline to endpoint visit in the Montgomery-Asberg Depression Rating Scale (MADRS) total score for both doses. The MADRS is a ten-item, clinician-rated scale used to assess severity of depressive symptoms. Scores on the MADRS range from 0 to 60, with higher scores indicating more severe depression. Clinical Global Impression – Severity (CGI-S) was evaluated in Studies 3 and 4. VIIBRYD 20 mg and 40 mg demonstrated superiority over placebo as measured by improvement in CGI-S score.
Table 5. Summary of Results for the Primary Efficacy Endpoint – MADRS Total Score:
| Study Number | Treatment Group | Number of Patientsa | Mean Baseline Score (SD) | LS Mean Change from Baseline (SE) | Placebo-subtracted Differenceb (95% CI) |
|---|---|---|---|---|---|
| Study 1 | VIIBRYD 40mg/day | 198 | 30.8 (3.90) | -12.9 (0.77) | -3.2 (-5.2, -1.3) |
| Placebo | 199 | 30.7 (3.93) | -9.6 (0.76) | ||
| Study 2 | VIIBRYD 40 mg/day | 231 | 31.9 (3.50) | -13.3 (0.90) | -2.5 (-4.4, -0.6) |
| Placebo | 232 | 32.0 (3.63) | -10.8 (0.90) | ||
| Study 3 | VIIBRYD 40 mg/day | 253 | 30.7 (3.3) | -16.1 (0.64) | -5.1 (-6.9, -3.3) |
| Placebo | 252 | 30.9 (3.3) | -11.0 (0.65) | ||
| Study 4 | VIIBRYD 20 mg/day* | 288 | 31.3 (3.5) | -17.3 (0.63) | -2.6 (-4.3, -0.8) |
| VIIBRYD 40 mg/day* | 284 | 31.2 (3.8) | -17.6 (0.65) | -2.8 (-4.6, -1.1) | |
| Placebo | 281 | 31.4 (3.8) | -14.8 (0.62) |
SD = standard deviation; SE = standard error; LS Mean = least-square mean; CI = confidence interval
a based on patients who took study medication and had baseline and postbaseline MADRS assessments
b difference (drug minus placebo) in least-square mean change from baseline to endpoint
* All VIIBRYD treatment dose groups remained statistically significant compared with placebo after adjusting for multiplicity
Baseline demographics information were generally similar across all treatment groups. Examination of population subgroups based on age (there were few patients over 65), gender and race did not reveal any clear evidence of differential responsiveness.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.