VYVGART Solution for injection Ref.[116200] Active ingredients: Efgartigimod alfa
Source: European Medicines Agency (EU) Revision Year: 2026 Publisher: argenx BV, Industriepark-Zwijnaarde 7, 9052 Gent, Belgium
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: Immunosuppressants, selective immunosuppressants
ATC code: L04AA58
Mechanism of action
Efgartigimod alfa is a human IgG1 antibody fragment engineered for increased affinity to the neonatal Fc Receptor (FcRn). Efgartigimod alfa binds to FcRn, resulting in a reduction in the levels of circulating IgG including pathogenic IgG autoantibodies. Efgartigimod alfa does not affect the levels of other immunoglobulins (IgA, IgD, IgE or IgM), and does not reduce those of albumin.
IgG autoantibodies are the underlying cause of the pathogenesis of IgG mediated autoimmune diseases.
In MG these impair neuromuscular transmission by binding to acetylcholine receptors (AChR), musclespecific tyrosine kinase (MuSK) or low density lipoprotein receptor-related protein 4 (LRP4).
In CIDP, several lines of evidence point to the key role of IgG autoantibodies in the pathogenesis of this disease. This includes the demonstration of autoreactive IgG antibodies against components of myelinated nerves, passive transfer of CIDP symptoms to animal models using sera or IgG's from patients with CIDP, and the therapeutic effect of plasma exchange and immunoadsorption for treating patients with CIDP.
Pharmacodynamic effects
Intravenous formulation
In the ARGX-113-1704 double-blind placebo-controlled study in gMG patients, efgartigimod alfa 10 mg/kg administered once weekly for 4 weeks decreased serum IgG levels and AChR autoantibody (AChRAb) levels. Maximum mean percentage decrease in total IgG levels compared to baseline reached 61% one week after the last infusion of the initial treatment cycle and returned to baseline levels 9 weeks after the last infusion. Similar effects were also observed for all subtypes of IgG. Decrease in AChR-Ab levels followed a similar time course with maximum mean percentage decrease of 58% one week after the last infusion and return to baseline levels 7 weeks after the last infusion. Similar changes were observed during the second cycle of the study.
Subcutaneous formulation
In the ARGX-113-2001 study, decreases in AChR-Ab levels followed a comparable time course as total IgG levels and were similar between the efgartigimod alfa subcutaneous and intravenous groups. Maximum mean percentage decreases in AChR-Ab levels of 62.2% and 59.6% were observed one week after the last administration in the efgartigimod alfa subcutaneous and intravenous groups, respectively. For both the efgartigimod alfa subcutaneous and intravenous groups, decrease in total IgG and AChR-Ab levels were associated with a clinical response, as measured by the change from baseline in MG-ADL total score (see figure 1).
Figure 1. Relationship between total IgG and AChR-Ab and MG-ADL total score in AChR-Ab seropositive population treated with efgartigimod alfa subcutaneous (1A) and efgartigimod alfa intravenous (1B) (study ARGX-113-2001):
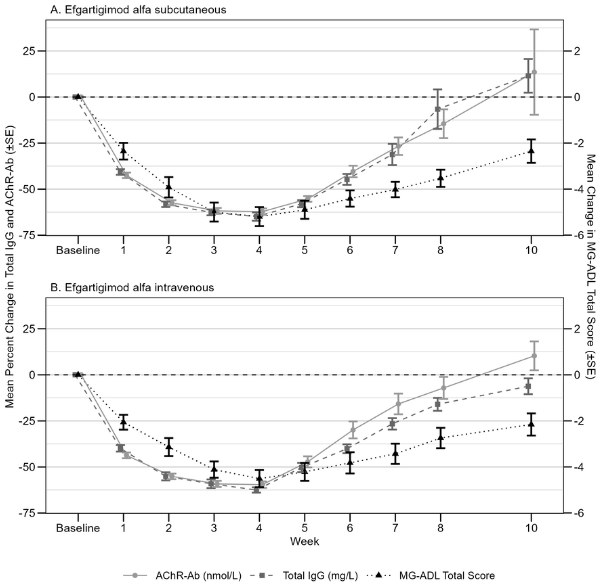
In the ARGX-113-1802 study in patients with CIDP receiving continuous once-weekly administration of efgartigimod alfa subcutaneous at 1 000 mg, the mean percent change from baseline in total IgG levels was sustained from Week 4 throughout the treatment period (mean percentage reduction from baseline ranging between 66.8 to 71.6%).
Clinical efficacy and safety
Generalised Myasthenia Gravis
Intravenous formulation
Efficacy of efgartigimod alfa for the treatment of adults with generalised Myasthenia Gravis (gMG) was studied in a 26-week, multicentre randomised double-blind placebo-controlled trial (ARGX-113-1704).
In this study, patients had to meet the following main criteria at screening:
- Myasthenia Gravis Foundation of America (MGFA) clinical classification class II, III or IV;
- Patients with either positive or negative serologic tests for antibodies to AChR;
- MG-Activities of Daily Living (MG-ADL) total score of ≥ 5;
- On stable doses of MG therapy prior to screening, that included acetylcholinesterase (AChE) inhibitors, steroids or non-steroidal immunosuppressive therapy (NSIST), either in combination or alone [NSISTs included but were not limited to azathioprine, methotrexate, cyclosporine, tacrolimus, mycophenolate mofetil, and cyclophosphamide];
- IgG levels of at least 6 g/L.
Patients with MGFA Class V gMG; patients with documented lack of clinical response to PLEX; patients treated with PLEX, IVIg one month and monoclonal antibodies six months prior to starting treatment; and patients with active (acute or chronic) hepatitis B infection, hepatitis C seropositivity, and diagnosis of AIDS, were excluded from the trials.
A total of 167 patients were enrolled in the study and were randomised to either efgartigimod alfa intravenous (n=84) or placebo (n=83). Baseline characteristics were similar between treatment groups, including median age at diagnosis [45 (19-81) years], gender [most were female; 75% (efgartigimod alfa) versus 66% (placebo)], race [most patients were white; 84.4%] and median time since diagnosis [8.2 years (efgartigimod alfa) and 6.9 years (placebo)].
The majority of patients (77% in each group) tested positive for antibodies to AChR (AChR-Ab) and 23% of patients tested negative for AChR-Ab.
During the study, over 80% of patients in each group received AChE inhibitors, over 70% in each treatment group received steroids, and approximately 60% in each treatment group received NSISTs, at stable doses. At study entry, approximately 30% of patients in each treatment group had no previous exposure to NSISTs.
Median MG-ADL total score was 9.0 in both treatment groups, and median Quantitative Myasthenia Gravis (QMG) total score was 17 and 16 in the efgartigimod alfa and placebo groups, respectively.
Patients were treated with efgartigimod alfa intravenous 10 mg/kg administered once weekly for 4 weeks and received a maximum of 3 treatment cycles.
The efficacy of efgartigimod alfa was measured using the Myasthenia Gravis-Specific Activities of Daily Living scale (MG-ADL) which assesses the impact of gMG on daily functions. A total score ranges from 0 to 24 with the higher scores indicating more impairment. In this study, an MG-ADL responder was a patient with ≥2-point reduction in the total MG-ADL score compared to the treatment cycle baseline, for at least 4 consecutive weeks with the first reduction occurring no later than 1 week after the last infusion of the cycle.
The efficacy of efgartigimod alfa was also measured using the QMG total score which is a grading system that assesses muscle weakness with a total possible score of 0 to 39 where higher scores indicate more severe impairment. In this study, a QMG responder was a patient who had a ≥3-point reduction in the total QMG score compared to the treatment cycle baseline, for at least 4 consecutive weeks with the first reduction occurring no later than 1 week after last infusion of the cycle.
The primary efficacy endpoint was the comparison of the percentage of MG-ADL responders during the first treatment cycle (C1) between treatment groups in the AChR-Ab seropositive population.
A key secondary endpoint was the comparison of the percentage of QMG responders during C1 between both treatment groups in the AChR-Ab seropositive patients.
Table 2. MG-ADL and QMG responders during cycle 1 in AChR-Ab seropositive population (mITT analysis set):
| Population | Efgartigimod alfa n/N (%) | Placebo n/N (%) | P-value | Difference Efgartigimod alfa- Placebo (95% CI) | |
|---|---|---|---|---|---|
| MG-ADL | AChR-Ab seropositive | 44/65 (67.7) | 19/64 (29.7) | <0.0001 | 38.0 (22.1; 54.0) |
| QMG | AChR-Ab seropositive | 41/65 (63.1) | 9/64 (14.1) | <0.0001 | 49.0 (34.5; 63.5) |
AChR-Ab = acetylcholine receptor-antibody; MG-ADL = Myasthenia Gravis Activities of Daily Living; QMG = Quantitative Myasthenia Gravis; mITT = modified intent-to-treat; n = number of patients for whom the observation was reported; N = number of patients in the analysis set; CI = confidence interval; Logistic regression stratified for AChR-Ab status (if applicable), Japanese/Non-Japanese and standard of care, with baseline MG-ADL as covariate/QMG as covariates
Two-sided exact p-value
Analyses show that during the second treatment cycle MG-ADL responder rates were similar to those during the first treatment cycle (see Table 3).
Table 3. MG-ADL and QMG responders during cycle 2 in AChR-Ab seropositive population (mITT analysis set):
| Population | Efgartigimod alfa n/N (%) | Placebo n/N (%) | |
|---|---|---|---|
| MG-ADL | AChR-Ab seropositive | 36/51 (70.6) | 11/43 (25.6) |
| QMG | AChR-Ab seropositive | 24/51 (47.1) | 5/43 (11.6) |
AChR-Ab = acetylcholine receptor-antibody; MG-ADL = Myasthenia Gravis Activities of Daily Living; QMG = Quantitative Myasthenia Gravis; mITT = modified intent-to-treat; n = number of patients for whom the observation was reported; N = number of patients in the analysis set.
Exploratory data shows that onset of response was observed within 2 weeks of initial infusion in 37/44 (84%) patients treated with efgartigimod alfa intravenous in the AChR-Ab seropositive MG-ADL responders.
In the double-blind placebo-controlled study (ARGX-113-1704), the earliest possible time to initiating the subsequent treatment cycle was 8 weeks after the initial infusion of the first treatment cycle. In the overall population the mean time to the second treatment cycle in the efgartigimod alfa intravenous group was 13 weeks (SD 5.5 weeks) and the median time was 10 weeks (8-26 weeks) from the initial infusion of the first treatment cycle. In the open-label extension study (ARGX-113-1705) the earliest possible time of initiation of the subsequent treatment cycles was 7 weeks.
In patients that responded to treatment, the duration of clinical improvement was 5 weeks in 5/44 (11%) patients, 6-7 weeks in 14/44 (32%) of patients, 8-11 weeks in 10/44 (23%) patients and 12 weeks or more in 15/44 (34%) patients.
Subcutaneous formulation
A 10-week, randomised, open-label, parallel-group, multicentre study (ARGX-113-2001) was conducted in adult patients with gMG to evaluate the non-inferiority of the pharmacodynamic effect of efgartigimod alfa subcutaneous compared to efgartigimod alfa intravenous. The main inclusion and exclusion criteria were the same as in study ARGX-113-1704.
A total of 110 patients were randomised and received one cycle of once weekly administrations for 4 weeks, of either efgartigimod alfa subcutaneous 1 000 mg (n=55) or efgartigimod alfa intravenous 10 mg/kg (n=55). The majority of patients were positive for antibodies to AChR (AChR-Ab): 45 patients (82%) in efgartigimod alfa subcutaneous group and 46 patients (84%) in efgartigimod alfa intravenous group. All patients were on stable doses of MG therapy prior to screening, that included AChE inhibitors, steroids or NSISTs, either in combination or alone.
Baseline characteristics were similar between treatment groups.
During the study, over 80% of patients in each group received AChE inhibitors, over 60% of patients in each group received steroids and about 40% in each treatment group received NSISTs, at stable doses. At study entry, approximately 56% of patients in each treatment group had no previous exposure to NSISTs.
The primary endpoint was the comparison of the percent reduction in total IgG levels from baseline at day 29 between treatment groups in the overall population. The results in the AChR-Ab seropositive population demonstrates non-inferiority of efgartigimod alfa subcutaneous compared to efgartigimod alfa intravenous (see Table 4).
Table 4. ANCOVA analysis of percent change from baseline in total IgG level at day 29 in AChR-Ab seropositive population (mITT analysis set):
| Efgartigimod alfa SC | Efgartigimod alfa IV | Difference Efgartigimod alfa SC- Efgartigimod alfa IV | ||||||
|---|---|---|---|---|---|---|---|---|
| N | LS Mean | 95% CI | N | LS Mean | 95% CI | LS of Mean difference | 95% CI | p-value |
| 41 | -66.9 | -69.78, -64.02 | 43 | -62.4 | -65.22, -59.59 | -4.5 | -8.53, -0.46 | <0.0001 |
AChR-Ab = acetylcholine receptor–antibody; ANCOVA = analysis of covariance; CI = confidence interval; SC = subcutaneous; IV = intravenous; LS = least squares; mITT = modified intent-to-treatment analysis set; N = number of patients per group that were included in the ANCOVA analysis
Efficacy secondary endpoints were comparisons of the percentage of MG-ADL and QMG responders, as defined in study ARGX-113-1704, between both treatment groups. The results in AChR-Ab seropositive population are presented in Table 5.
Table 5. MG-ADL and QMG responders at day 29 in AChR-Ab seropositive population (mITT analysis set):
| Efgartigimod alfa SC n/N (%) | Efgartigimod alfa IV n/N (%) | Difference Efgartigimod alfa SC- Efgartigimod alfa IV (95% CI) | |
|---|---|---|---|
| MG-ADL | 32/45 (71.1) | 33/46 (71.7) | -0.6 (-19.2 to 17.9) |
| QMG | 31/45 (68.9) | 24/45 (53.3) | 15.6 (-4.3 to 35.4) |
AChR-Ab = acetylcholine receptor-antibody; MG-ADL = Myasthenia Gravis Activities of Daily Living; QMG = Quantitative Myasthenia Gravis; SC = subcutaneous; IV = intravenous; mITT = modified intent-to-treat; n = number of patients for whom the observation was reported; N = number of patients in the analysis set; CI = confidence interval
Exploratory data shows that onset of response was observed within 2 weeks of initial administration in 28/32 (88%) patients treated with efgartigimod alfa subcutaneous and 27/33 (82%) patients treated with efgartigimod alfa intravenous in the AChR‑Ab seropositive MG-ADL responders.
Chronic Inflammatory Demyelinating Polyneuropathy
The efficacy of efgartigimod alfa subcutaneous for the treatment of adults with CIDP was studied in a prospective, multicentre study ARGX-113-1802 conducted in 2 treatment stages: an open-label Stage A and a randomized-withdrawal, double-blinded, placebo-controlled Stage B.
Patients had been either on or off CIDP treatment during the 6 months prior to study entry. Those on prior CIDP treatment as well as those off CIDP treatment with no documented evidence of recent CIDP deterioration, entered a treatment-free run-in period, and patients who demonstrated evidence of clinically meaningful deterioration then entered Stage A of the study. Those off CIDP treatment who had recent documented evidence of CIDP deterioration, skipped the run-in period and entered straight into Stage A.
A total of 322 patients were enrolled in Stage A. Patients received up to 12 once weekly injections of efgartigimod alfa subcutaneous 1 000 mg until evidence of clinical improvement (ECI) occurred at 2 consecutive study visits. Subsequently, the patients with confirmed ECI entered Stage B of the study and were randomised to receive weekly administrations of either efgartigimod alfa subcutaneous (111 patients) or placebo (110 patients). ECI was defined as clinical improvement on adjusted Inflammatory Neuropathy Cause and Treatment (aINCAT) or improvement on Inflammatory Rasch- built Overall Disability Scale (I-RODS)/Grip Strength in patients who deteriorated on these scales only prior to Stage A.
In Stage A, patients had a median age of 54 years (range: 20 to 82 years), a median time since CIDP diagnosis of 2.8 years and median INCAT score of 4.0. Sixty-five percent were male and 66% were White. In Stage B, patients had a median age of 55 years (range: 20 to 82 years), a median time since CIDP diagnosis of 2.2 years and median INCAT score of 3.0. Sixty-four percent were male and 65% were White. Baseline characteristics of Stage B were similar between treatment groups.
In Stage A, the primary endpoint was the percentage of responders defined as patients achieving confirmed ECI. The primary endpoint was met in 66.5% of patients; further details are presented in Table 6.
A secondary endpoint in Stage A was the time to the first confirmed ECI. Week 4 was the earliest time point at which ECI criteria could be met. At that time point, up to 40% of patients achieved ECI. Based on an additional pre-specified analysis, 25% of patients showed clinically relevant improvement after 9 days in at least one of 3 parameters (aINCAT, I-RODS or Grip Strength).
The majority of patients achieved confirmed ECI across all prior CIDP medication groups.
Table 6. Evidence of clinical improvement in patients with CIDP in ARGX-113-1802 Stage A:
| ECI responders and time to initial confirmed ECI | Stage A |
|---|---|
| Efgartigimod alfa SC (N=322) | |
| ECI Responders (patients with confirmed clinical improvement) n/N (%) (95% CI) | 214/322 (66.5%) (61.0; 71.6) |
| Time to initial confirmed ECI in days median (95% CI) | 43.0 (31.0; 51.0) |
n = number of patients for whom the observation was reported; N = number of patients in the analysis set
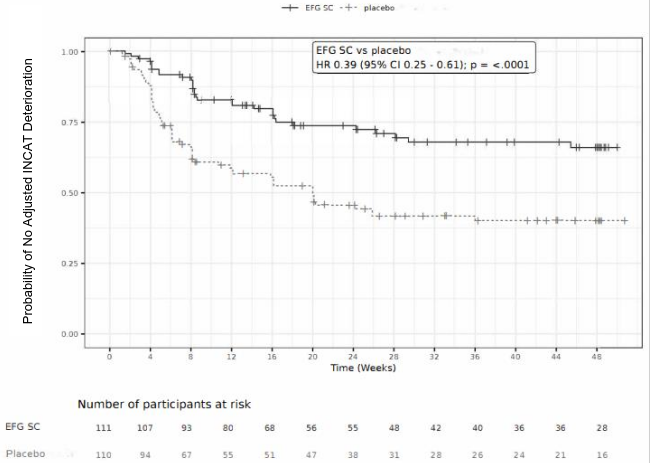
In Stage B, the primary endpoint was defined as the time to the occurrence of the first evidence of clinical deterioration (a 1-point increase in aINCAT compared to Stage B baseline, which was confirmed at a consecutive visit after the first 1-point increase in aINCAT or, a ≥2-point increase in aINCAT compared to Stage B baseline). Patients who received efgartigimod alfa subcutaneous remained relapse-free (i.e., no clinical deterioration) significantly longer compared to patients who received placebo, as demonstrated by a hazard ratio of 0.394 [95% CI (0.253; 0.614)]. 31/111 (27.9%) of patients who received efgartigimod alfa subcutaneous during Stage B of the study relapsed compared to 59/110 (53.6%) of patients who received placebo. The results are presented in Table 7 and Figure 2.
Table 7. First evidence of clinical deterioration in patients with CIDP in study ARGX-113-1802 Stage B:
| Time to 1st aINCAT increase (clinical deterioration) | Stage B | |
|---|---|---|
| Efgartigimod alfa SC (N=111) | Placebo (N=110) | |
| Hazard ratio (95% CI) | 0.394 (0.253; 0.614) p-value < 0.0001 | |
| Median time in days (95% CI) | NC (NC; NC) | 140.0 (75.0; NC) |
NC = not calculated; N = number of patients in the analysis set; aINCAT = adjusted Inflammatory Neuropathy Cause and Treatment
Figure 2. Time to the first aINCAT deterioration (Kaplan-Meier Curve) in patients with CIDP in study ARGX-113-1802 Stage B:
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with Vyvgart in one or more subsets of the paediatric population in treatment of myasthenia gravis (see section 4.2 for information on paediatric use).
The European Medicines Agency has waived the obligation to submit the results of studies with Vyvgart in all subsets of the paediatric population in CIDP (see section 4.2 for information on paediatric use).
5.2. Pharmacokinetic properties
Absorption
Based upon population PK data analysis, the estimated bioavailability with efgartigimod alfa 1 000 mg subcutaneous is 77%.
The mean Ctrough after 4 once weekly administrations with efgartigimod alfa 1 000 mg subcutaneous and efgartigimod alfa 10 mg/kg intravenous were 22.0 μg/mL (37% CV) and 14.9 μg/mL (43% CV), respectively. The AUC0-168h of efgartigimod alfa after administration of one treatment cycle with 1 000 mg subcutaneous and 10 mg/kg intravenous were comparable.
In patients receiving continuous subcutaneous administration of efgartigimod alfa 1 000 mg once weekly, mean Ctrough ranged from 14.9 to 20.1 μg/mL.
Distribution
Based upon population PK data analysis in healthy subjects and patients the volume of distribution is 18 L.
Biotransformation
Efgartigimod alfa is expected to be degraded by proteolytic enzymes into small peptides and amino acids.
Elimination
The terminal half-life is 80 to 120 hours (3 to 5 days). Based upon population PK data analysis, the clearance is 0.128 L/h. The molecular weight of efgartigimod alfa is approximately 54 kDa, which is at the boundary of molecules that are renally filtered.
Linearity/non-linearity
The pharmacokinetics profile of efgartigimod alfa is linear, independent of dose or time, with minimal accumulation.
Special populations
Age, gender, race and bodyweight
The pharmacokinetics of efgartigimod alfa were not affected by age (19-84 years), gender, race and bodyweight.
Renal impairment
No dedicated pharmacokinetic studies have been performed in patients with renal impairment.
The effect of renal function marker estimated glomerular filtration rate [eGFR] as a covariate in a population pharmacokinetic analysis showed an increase in exposure (11% to 21%) in patients with mild renal impairment (eGFR 60-89 mL/min/1.73 m²). No specific dose adjustment is recommended in patients with mild renal impairment.
There is insufficient data on the impact of moderate renal impairment (eGFR 30-59 mL/min/1.73 m²) and severe renal impairment (eGFR <30 mL/min/1.73 m²) on efgartigimod alfa pharmacokinetic parameters.
Hepatic impairment
No dedicated pharmacokinetic study has been performed in patients with hepatic impairment.
The effect of hepatic function markers as covariates in a population pharmacokinetic analysis did not show any impact on the pharmacokinetics of efgartigimod alfa.
5.3. Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology and repeated dose toxicity.
In reproduction studies in rats and rabbits, intravenous administration of efgartigimod alfa did not result in adverse effects on fertility and pregnancy nor were teratogenic effects observed up to dose levels corresponding to 11-fold (rats) and 56-fold (rabbits) a human 10 mg/kg exposure based on AUC.
Carcinogenicity and genotoxicity
No studies have been conducted to assess the carcinogenic and genotoxic potential of efgartigimod alfa.
Hyaluronidase is found in most tissues of the human body. Non-clinical data for recombinant human hyaluronidase reveal no special hazard for humans based on conventional studies of repeated dose toxicity including safety pharmacology endpoints. Reproductive toxicology studies with rHuPH20 revealed embryofoetal toxicity in mice at high systemic exposure, but did not show teratogenic potential.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.