WAKIX Film-coated tablet Ref.[7629] Active ingredients: Pitolisant
Source: European Medicines Agency (EU) Revision Year: 2025 Publisher: Bioprojet Pharma, 9, rue Rameau, 75002 Paris, France, Tel: +33 (0)1 47 03 66 33, Fax: +33 (0)1 47 03 66 30, e-mail: contact@bioprojet.com
Pharmacodynamic properties
Pharmacotherapeutic group: Other nervous system drugs
ATC code: N07XX11
Mechanism of action
Pitolisant is a potent, orally active histamine H3-receptor antagonist/inverse agonist which, via its blockade of histamine auto-receptors enhances the activity of brain histaminergic neurons, a major arousal system with widespread projections to the whole brain. Pitolisant also modulates various neurotransmitter systems, increasing acetylcholine, noradrenaline and dopamine release in the brain. However no increase in dopamine release in the striatal complex including nucleus accumbens was evidenced for pitolisant.
Pharmacodynamic effects
In narcoleptic patients with or without cataplexy, pitolisant improves the level and duration of wakefulness and daytime alertness assessed by objective measures of ability to sustain wakefulness (e.g. Maintenance of Wakefulness Test (MWT)) and attention (e.g. Sustained Attention to Response Task (SART)).
Clinical efficacy and safety
Adult population
Narcolepsy (with or without cataplexy) is a chronic condition. The effectiveness of pitolisant up to 36 mg once a day, for the treatment of narcolepsy with or without cataplexy was established in two main, 8 weeks, multicenter, randomized, double-blind, placebo-controlled, parallel group trials (Harmony I and Harmony CTP). Harmony Ibis, study with a similar design, was limited to 18 mg once a day. Long-term safety data of Wakix in this indication are available in the open label long-term study HARMONY III.
The pivotal study (Harmony 1), double-blind, randomized, vs placebo and modafinil (400 mg/day), parallel group studies with flexible dose adaptation, included 94 patients (31 patients treated with pitolisant, 30 with placebo and 33 with modafinil). Dosage was initiated at 9 mg once a day and was increased, according to efficacy response and tolerance to 18 mg or 36 mg once a day per 1-week interval. Most patients (60%) reached the 36 mg once a day dosage. To assess the efficacy of pitolisant on Excessive Daytime Sleepiness (EDS), Epworth Sleepiness Scale (ESS) score was used as primary efficacy criterion. The results with pitolisant were significantly superior to those in the placebo group (mean difference: -3.33; 95%CI [-5.83 to -0.83]; p<0.05) but did not differ significantly from the results in the modafinil group (mean difference: 0.12; 95%CI [-2.5 to 2.7]). The waking effect of the two active substances was established at similar rates (Figure 1).
Figure 1. Changes in Epworth Sleepiness Scale Score (ESS) (mean ± SEM) from Baseline to week 8 in Harmony 1 study:
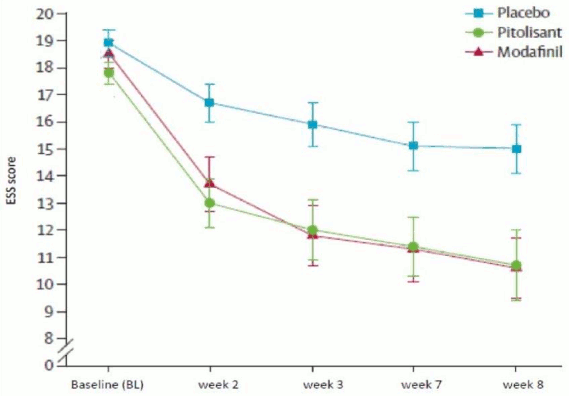
The effect on Epworth was supported in two laboratory tests of vigilance and attention (Maintenance of Wakefulness Test (MWT) (p=0.044) and Sustained Attention to Response (SART) (p=0.053, almost but not significant)).
Cataplexy attacks frequency in patients displaying this symptom was decreased significantly (p=0.034) with pitolisant (-65%) compared to placebo (-10%). The daily cataplexy rate (geometric means) was 0.52 at baseline and 0.18 at final visit for pitolisant and 0.43 at baseline and 0.39 at final visit for placebo, with a rate ratio rR=0.38 [0.16; 0.93] (p=0.034).
The second pivotal study (Harmony Ibis) included 165 patients (67 treated with pitolisant, 33 with placebo and 65 with modafinil). The study design was similar to study Harmony I except that the maximum dose for pitolisant reached by 75% of patients was 18 mg once a day instead of 36 mg in Harmony I. As an important unbalance led to comparison of results with or without cluster grouping of sites, the most conservative approach showed non-significant ESS score decrease with pitolisant compared to placebo (pitolisant-placebo=-1.94 with p=0.065). Results from cataplexy rate at 18 mg once a day were not consistent with those of the first pivotal study (36 mg once a day).
Improvement of the two objective tests of wakefulness and attention, MWT and SART, with pitolisant was significant versus placebo (p=0.009 and p=0.002 respectively) and non-significant versus modafinil (p=0.713 and p=0.294 respectively).
Harmony CTP, a supportive double blind, randomized, parallel group study of pitolisant versus placebo, was designed to establish pitolisant efficacy in patients with high frequency cataplexy in narcolepsy. The primary efficacy endpoint was the change in the average number of cataplexy attacks per week between the 2 weeks of baseline and the 4 weeks of stable treatment period at the end of study. 105 narcoleptic patients with high frequency weekly cataplexy rates at baseline were included (54 patients treated with pitolisant and 51 with placebo). Dosage was initiated at 4.5 mg once a day and was increased, according to efficacy response and tolerance to 9 mg, 18 mg or 36 mg once a day per 1-week interval. Most patients (65%) reached the 36 mg once a day dosage.
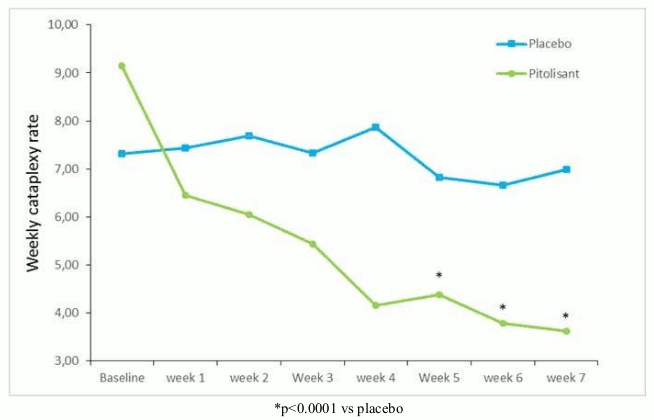
On the primary efficacy endpoint, Weekly Rate of Cataplexy episodes (WRC), the results with pitolisant were significantly superior to those in the placebo group (p<0.0001), with a progressive 64% decrease from baseline to end of treatment (Figure 2). At baseline, the geometric mean of WRC was 7.31 (median=6.5 [4.5; 12]) and 9.15 (median=8.5 [5.5; 15.5]) in the placebo and pitolisant groups respectively. During the stable period (until the end of treatment), geometric mean WRC decreased to 6.79 (median=6 [3; 15]) and 3.28 (median=3 [1.3; 6]) in the placebo and pitolisant groups respectively in patients who had experienced at least one episode of cataplexy. The observed WRC in pitolisant group was about half of WRC in the placebo group: the effect size of pitolisant compared with placebo was summarized by the ratio rate rR(Pt/Pb), rR=0.512; 95%CI [0.435 to 0.603]; p<0.0001). The effect size of pitolisant compared with placebo based on a model for WRC based on BOCF with centre as a fixed effect was 0.581, 95%CI [0.493 to 0.686]; p<0.0001.
Figure 2. Changes in weekly cataplexy episodes (geometric mean) from Baseline to week 7 in Harmony CTP study:
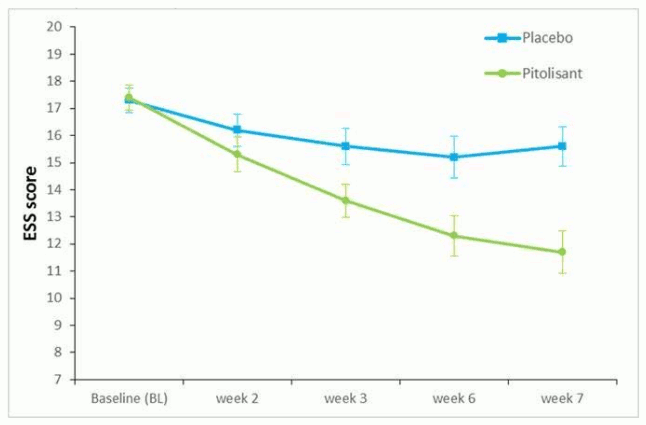
The effect of pitolisant on EDS was also assessed in this population using the ESS score. In the pitolisant group, ESS decreased significantly between baseline and the end of treatment compared to placebo with an observed mean change of -1.9 ± 4.3 and -5.4 ± 4.3 (mean ± sd) for placebo and pitolisant respectively, (p<0.0001) (Figure 3). This effect on EDS was confirmed by the results on Maintenance of Wakefulness Test (MWT). The geometric mean of the ratios (MWT Final/MWT Baseline) was 1.8 (95%CI 1.19; 2.71, p=0.005). The MWT value in the pitolisant group was 80% higher than in the placebo group.
Figure 3. Changes in Epworth Sleepiness Scale Score (ESS) (mean ± SEM) from Baseline to week 7 in Harmony CTP study:
The open-label, long-term Phase III study (HARMONY III) assessed the long term safety of pitolisant in patients suffering from narcolepsy (with or without cataplexy) over 12 months and with an extension of up to 5 years. 102 narcoleptic patients with or without cataplexy were included in the 12 months follow-up period. 68 patients completed the first 12 months period. 45, 38, 34 and 14 patients completed the 2, 3, 4 and 5 year follow-up periods, respectively.
The maximal dose received during the study was 36 mg/day in 85% of patients. After 12 months of treatment, improvements in EDS assessed by ESS score of remaining patients is of same magnitude as those observed in the other trials conducted in narcoleptic patients. The decrease in mean ESS score (SD) was -3.62 (4.63) after 1 year.
After 12 months of treatment with pitolisant, frequency of symptoms such as sleep attacks, sleep paralysis, cataplexy and hallucinations has been improved.
No major safety concern was identified. The safety results observed were similar to those reported in previous trials where pitolisant at 36 mg once daily was given for up to 3 months only.
Paediatric population
The effectiveness of pitolisant up to 36 mg once a day has been studied for the treatment of narcolepsy with or without cataplexy in children from 6 to less than 18 years old in an 8-week, multicenter, randomized, double-blind, placebo-controlled, parallel group trial. It included 110 patients (72 patients in the pitolisant group, 38 in the placebo group). Dosage was initiated at 4.5 mg once a day and was increased, according to efficacy response and tolerance to 18 mg or 36 mg once a day per 1-week interval. Patients weighing less than 40 kg remained at a maximum dose of 18 mg. Most patients (60%) reached the 36 mg once a day dosage. 35 patients (31.8%) were aged 6 to 11 years and 75 patients (68.2%) were aged 12 to less than 18 years. To assess the efficacy of pitolisant on Excessive Daytime Sleepiness (EDS) and cataplexy (CTP), the Ullanlinna Narcolepsy Scale (UNS) total score was used as primary efficacy criterion, assessed as the change from baseline to the end of double-blind period. The estimate LS means difference (SE) [95% CI] of UNS between treatment groups (pitolisant minus placebo) was -3.69 (1.37) [-6.38; -0.99], p=0.0073. Secondary endpoints included the paediatric daytime sleepiness scale (PDSS), the UNS-cataplexy (CTP) subscore, and the weekly rate of cataplexy (WRC). The estimate LS means difference (SE) [95% CI] of the PDSS total score between treatment groups (pitolisant minus placebo) was -3.41 (1.07) [-5.52; -1.31], p=0.0015. In the subgroup of patients with type 1 narcolepsy, who had no minimum level of cataplexy required at inclusion (N=61 in the pitolisant group; N=29 in the placebo group), the estimate LS means difference (SE) [95% CI] of the UNS-CTP subscore between treatment groups (pitolisant minus placebo) was -1.77 (0.78) [-3.29; -0.24], p=0.0229, and the rate ratio between the WRC in the pitolisant group and the WRC in the placebo group, adjusted for baseline, was in favor of pitolisant (0.42 [95% CI: 0.18; 1.01], p=0.0540).
Table 1. overview of efficacy results after 8 weeks in phase 3 paediatric study:
| Placebo (n=38) | Pitolisant (n=72) | |
| Ullanlinna Narcolepsy Scale (UNS) | ||
| Total score | ||
| Baseline mean (SD) End of treatment mean (SD) LS mean (SE) – change from baseline Estimate, 95% CI p-value | 23.68 (9.08) 21.77 (9.25) -2.60 (1.35) | 24.63 (7.80) 18.23 (8.14) -6.29 (1.14) -3.69 (-6.38; -0.99) 0.0073 |
| Paediatric Daytime Sleepiness Score | ||
| Baseline mean (SD) End of treatment mean (SD) LS mean (SE) – change from baseline Estimate, 95% CI p-value | 20.00 (3.49) 17.96 (5.60) -2.11 (0.89) | 20.16 (3.64) 14.57 (5.37) -5.53 (0.66) -3.41 (-5.52; -1.31) 0.0015 |
| Placebo (n=29) | Pitolisant (n=61) | |
| UNS-Cataplexy Subscore* | ||
| Baseline mean (SD) End of treatment mean (SD) LS mean (SE) – change from baseline Estimate, 95% CI p-value | 9.03 (4.33) 8.07 (4.62) -1.12 (0.64) | 8.93 (3.96) 6.02 (4.00) -2.88 (0.44) -1.77 (-3.29; -0.24) 0.0229 |
| Weekly cataplexy rate* | ||
| Baseline mean (SD) LS mean (SE) Estimate, 95% CI p-value | 13.44 (26.92) 5.05 (0.37) | 8.63 (17.73) 2.14 (0.27) 0.42 (0.18; 1.01) 0.0540 |
* only measured in patients with type I narcolepsy
Figure 4. Change in the Mean Ullanlinna Narcolepsy Scale Total Score (mean ± SEM) from Baseline to the End of Treatment (Full Analysis Set):
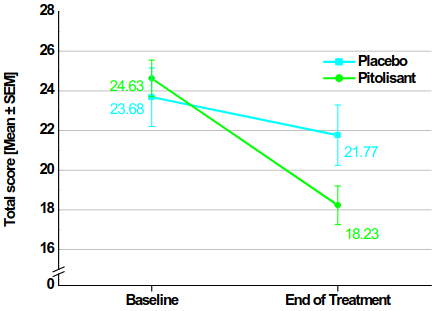
Baseline=[V1 score (D-14) + V2 score (D0)]/2
End of treatment=[V6 score (D49) + V7 score (D56)]/2
SEM=standard error of the mean
Pharmacokinetic properties
The exposure to pitolisant in healthy volunteers was assessed in studies involving more than 200 subjects that received doses of pitolisant in single administration up to 216 mg and for a duration up to 28 days.
Absorption
Pitolisant is well and rapidly absorbed with peak plasma concentration reached approximately three hours after administration.
Distribution
Pitolisant exhibits high serum protein binding (>90%) and demonstrates approximately equal distribution between red blood cells and plasma.
Biotransformation
The metabolisation of pitolisant in humans is fully characterized. The major non-conjugated metabolites are hydroxylated derivatives in several positions and cleaved forms of pitolisant leading to inactive major carboxylic acid metabolite found in urine and serum. They are formed under the action of CYP3A4 and CYP2D6. Several conjugated metabolites were identified, the major ones (inactive) being two glycine conjugates of the acid metabolite of pitolisant and a glucuronide of a ketone metabolite of monohydroxy desaturated pitolisant.
On liver microsomes, pitolisant and its major metabolites do not significantly inhibit the activities of the cytochromes CYP1A2, CYP2C9, CYP2C19, CYP2C8, CYP2B6, CYP2E1 or CYP3A4 and of uridine diphosphate glucuronosyl transferases isoforms UGT1A1, UGT1A4, UGT1A6, UGT1A9 and UGT2B7 up to the concentration of 13.3 µM, a level considerably higher than the levels achieved with therapeutic dose. Pitolisant is an inhibitor of CYP2D6 with moderate potency (IC50 = 2.6 µM).
Pitolisant induces CYP3A4, CYP1A2 and CYP2B6 in vitro. Clinically relevant interactions are expected with CYP3A4 and CYP2B6 substrates and by extrapolation, UGTs, CYP2C and P-gp substrates (see section 4.5).
In vitro studies indicate that pitolisant is neither a substrate nor an inhibitor of human P-glycoprotein and breast cancer resistance protein (BCRP). Pitolisant is not a substrate of OATP1B1, OATP1B3. Pitolisant is not a significant inhibitor of OAT1, OAT3, OCT2, OATP1B1, OATP1B3, MATE1, or MATE2K at the tested concentration. Pitolisant shows greater than 50% inhibition towards OCT1 (organic cation transporters 1) at 1.33 µM, the extrapolated IC50 of pitolisant is 0.795 µM (see section 4.5).
Elimination
Pitolisant has a plasma half-life of 10-12 hours. Upon repeated administrations, the steady state is achieved after 5-6 days of administration leading to an increased serum level around 100%. Inter individual variability is rather high, some volunteers showing outlier high profile (without tolerance issues).
The elimination is mainly achieved via urine (approximately 63%) through an inactive non conjugated metabolite (BP2.951) and a glycine conjugated metabolite. 25% of the dose is excreted through expired air and a small fraction (<3%) recovered in faeces where the amount of pitolisant or BP2.951 was negligible.
Linearity/non-linearity
When pitolisant dose is doubled from 27 to 54 mg, AUC0-∞ is increased by about 2.3.
Special populations
Elderly
In 68 to 80 years old patients the pharmacokinetics of pitolisant is not different compared to younger patients (18 to 45 years of age). Above 80 years old, kinetics show a slight variation without clinical relevance. Limited data are available in elderly. Therefore, dosing should be adjusted according to their renal hepatic status (see section 4.2 and 4.4).
Renal impairment
In patients with impaired renal function (stages 2 to 4 according to the international classification of chronic kidney disease, i.e. creatinine clearance between 15 and 89 ml/min), Cmax and AUC tended to be increased by a factor of 2.5 without any impact on half-life (see section 4.2).
Hepatic impairment
In patients with mild hepatic impairment (Child-Pugh A), there was no significant changes in pharmacokinetics compared with normal healthy volunteers. In patients with moderate hepatic impairment (Child-Pugh B), AUC increased by a factor 2.4, while half-life doubled (see section 4.2). Pitolisant pharmacokinetics after repeated administration in patients with hepatic impairment has not been evaluated yet.
CYP2D6 poor metabolizers
The exposure to Pitolisant was higher in the CYP2D6 poor metabolisers after a single dose and at steady state; Cmax and AUC(0-tau) was approximately 2.7-fold and 3.2-fold greater on Day 1 and 2.1-fold and 2.4-fold on Day 7. The serum Pitolisant half-life was longer in CYP2D6 poor metabolisers compared to the extensive metabolisers.
Race
The effect of race on metabolism of pitolisant has not been evaluated.
Paediatric population
The pharmacokinetics of pitolisant at the dose of 18 mg in children from 6 to less than 18 years with narcolepsy has been studied in a multi-centre, single dose trial. By comparison to adult patients exposure, in a Population PK analysis with a body weight-dependent model, systemic exposure to pitolisant at the dose of 18 mg as estimated by Cmax and AUC0-10h are roughly 3-fold higher in children with a body weight below 40 kg and 2-fold higher in adolescents with a body weight above 40 kg compared to adults. Therefore, the dose titration should be initiated at the lowest dose of 4.5 mg and limited to 18 mg in children weighing less than 40 kg (see section 4.2).
Preclinical safety data
After 1 month in mice, 6 months in rats and 9 months in monkeys, no adverse effect level (NOAEL) were 75, 30 and 12 mg/kg/day, p.o., respectively, providing safety margins of 9, 1 and 0.4, respectively when compared to the drug exposure at therapeutic dose in human. In rats, transient reversible convulsive episodes occurred at Tmax , that may be attributable to a metabolite abundant in this species but not in humans. In monkeys, at the highest doses, transient CNS related clinical signs including emesis, tremors and convulsions were reported. At the highest doses, no histopathological changes were recorded in monkeys and rats presented some limited histopathological changes in some organs (liver, duodenum, thymus, adrenal gland and lung).
Pitolisant was neither genotoxic nor carcinogenic.
Teratogenic effect of pitolisant was observed at maternally toxic doses (teratogenicity safety margins <1 in rats and in rabbits). At high doses, pitolisant induced sperm morphology abnormalities and decreased motility without any significant effect on fertility indexes in male rats and it decreased the percentage of live conceptuses and increased post-implantation loss in female rats (safety margin of 1). It caused a delay in post-natal development (safety margin of 1).
Pitolisant/metabolites were shown to cross the placenta barrier in animals.
Juvenile toxicity studies in rats revealed that the administration of pitolisant at high doses induced a dose related mortality and convulsive episode that may be attributable to a metabolite abundant in rats but not in humans.
Pitolisant blocked hERG channel with an IC50 exceeding therapeutic concentrations and induced a slight QTc prolongation in dogs.
In preclinical studies, drug dependence and drug abuse liability studies were conducted in mice, monkeys and rats. However, no definitive conclusion could be drawn on tolerance, dependence and self-administration studies.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.