XIFAXAN Tablet Ref.[11129] Active ingredients: Rifaximin
Source: FDA, National Drug Code (US) Revision Year: 2020
12.1. Mechanism of Action
Rifaximin is an antibacterial drug [see Clinical Pharmacology (12.4)].
12.3. Pharmacokinetics
Absorption
In healthy subjects, the mean time to reach peak rifaximin plasma concentrations was about an hour and the mean Cmax ranged 2.4 to 4 ng/mL after a single dose and multiple doses of XIFAXAN 550 mg.
Travelers' Diarrhea
Systemic absorption of XIFAXAN (200 mg three times daily) was evaluated in 13 subjects challenged with shigellosis on Days 1 and 3 of a three-day course of treatment. Rifaximin plasma concentrations and exposures were low and variable. There was no evidence of accumulation of rifaximin following repeated administration for 3 days (9 doses). Peak plasma rifaximin concentrations after 3 and 9 consecutive doses ranged from 0.81 to 3.4 ng/mL on Day 1 and 0.68 to 2.26 ng/mL on Day 3. Similarly, AUC0-last estimates were 6.95 ± 5.15 ng•h/mL on Day 1 and 7.83 ± 4.94 ng•h/mL on Day 3. XIFAXAN is not suitable for treating systemic bacterial infections because of limited systemic exposure after oral administration [see Warnings and Precautions (5.1)].
Hepatic Encephalopathy
Mean rifaximin exposure (AUCτ) in patients with a history of HE was approximately 12-fold higher than that observed in healthy subjects. Among patients with a history of HE, the mean AUC in patients with Child-Pugh Class C hepatic impairment was 2-fold higher than in patients with Child-Pugh Class A hepatic impairment [see Warnings and Precautions (5.4), Use in Specific Populations (8.7)].
Irritable Bowel Syndrome with Diarrhea
In patients with irritable bowel syndrome with diarrhea (IBS-D) treated with XIFAXAN 550 mg three times a day for 14 days, the median Tmax was 1 hour and mean Cmax and AUC were generally comparable with those in healthy subjects. After multiple doses, AUCtau was 1.65-fold higher than that on Day 1 in IBS-D patients (Table 2).
Table 2. Mean (± SD) Pharmacokinetic Parameters of Rifaximin Following XIFAXAN 550 mg Three Times a Day in IBS-D Patients and Healthy Subjects:
| Healthy Subjects | IBS-D Patients | |||
|---|---|---|---|---|
| Single-Dose (Day 1) n=12 | Multiple-Dose (Day 14) n=14 | Single-Dose (Day 1) n=24 | Multiple-Dose (Day 14) n=24 | |
| Cmax (ng/mL) | 4.04 (1.51) | 2.39 (1.28) | 3.49 (1.36) | 4.22 (2.66) |
| Tmax (h)a | 0.75 (0.5-2.1) | 1.00 (0.5-2.0) | 0.78 (0-2) | 1.00 (0.5-2) |
| AUCtau (ng•h/mL) | 10.4 (3.47) | 9.30 (2.7) | 9.69 (4.16) | 16.0 (9.59) |
| Half-life (h) | 1.83 (1.38) | 5.63 (5.27) | 3.14 (1.71) | 6.08 (1.68) |
a Median (range)
Food Effect in Healthy Subjects
A high-fat meal consumed 30 minutes prior to XIFAXAN dosing in healthy subjects delayed the mean time to peak plasma concentration from 0.75 to 1.5 hours and increased the systemic exposure (AUC) of rifaximin by 2-fold but did not significantly affect Cmax.
Dose-Proportionality
After oral administration of XIFAXAN 200 mg, 400 mg, or 600 mg, rifaximin systemic exposures increased dose-dependently by approximately 2-fold for both AUClast and Cmax from 200 mg to 400 mg, but with a less than dose-proportional increase of 1.3-fold for both AUClast and Cmax from 400 mg to 600 mg administration.
Distribution
Rifaximin is moderately bound to human plasma proteins. In vivo, the mean protein binding ratio was 67.5% in healthy subjects and 62% in patients with hepatic impairment when XIFAXAN was administered.
Elimination
The mean half-life of rifaximin in healthy subjects at steady-state was 5.6 hours and was 6 hours in IBS-D patients.
Metabolism
In an in vitro study, rifaximin was metabolized mainly by CYP3A4. Rifaximin accounted for 18% of radioactivity in plasma suggesting that the absorbed rifaximin undergoes extensive metabolism.
Excretion
In a mass balance study, after administration of 400 mg 14C-rifaximin orally to healthy volunteers, of the 96.94% total recovery, 96.62% of the administered radioactivity was recovered in feces mostly as the unchanged drug and 0.32% was recovered in urine mostly as metabolites with 0.03% as the unchanged drug.
Biliary excretion of rifaximin was suggested by a separate study in which rifaximin was detected in the bile after cholecystectomy in patients with intact gastrointestinal mucosa.
Specific Populations
Hepatic Impairment
The systemic exposure of rifaximin was markedly elevated in patients with hepatic impairment compared to healthy subjects.
The pharmacokinetics of rifaximin in patients with a history of HE was evaluated after administration of XIFAXAN 550 mg twice a day. The pharmacokinetic parameters were associated with a high variability and mean rifaximin exposure (AUCτ) in patients with a history of HE was higher compared to those in healthy subjects. The mean AUCτ in patients with hepatic impairment of Child-Pugh Class A, B, and C was 10-, 14-, and 21-fold higher, respectively, compared to that in healthy subjects (Table 3).
Table 3. Mean (± SD) Pharmacokinetic Parameters of Rifaximin at Steady-State in Patients with a History of Hepatic Encephalopathy by Child-Pugh Class1:
| Healthy Subjects (n=14) | Child-Pugh Class | |||
|---|---|---|---|---|
| A (n=18) | B (n=15) | C (n=6) | ||
| AUCtau (ng•h/mL) | 12.3 ± 4.8 | 118 ± 67.8 | 169 ± 55.7 | 257 ± 100.2 |
| Cmax (ng/mL) | 3.4 ± 1.6 | 19.5 ± 11.4 | 25.4 ± 11.9 | 39.7 ± 13.4 |
| Tmax² (h) | 0.8 (0.5, 4.0) | 1 (0.9, 10) | 1 (1.0, 4.2) | 1 (0, 2) |
Renal Impairment
The pharmacokinetics of rifaximin in patients with impaired renal function has not been studied.
Drug Interaction Studies
An in vitro study suggests that rifaximin is a substrate of CYP3A4.
In vitro rifaximin is a substrate of P-glycoprotein, OATP1A2, OATP1B1, and OATP1B3. Rifaximin is not a substrate of OATP2B1.
Cyclosporine
In vitro in the presence of P-glycoprotein inhibitor, verapamil, the efflux ratio of rifaximin was reduced greater than 50%. In a clinical drug interaction study, mean Cmax for rifaximin was increased 83-fold, from 0.48 to 40.0 ng/mL; mean AUC∞ was increased 124-fold, from 2.54 to 314 ng•h/mL following co-administration of a single dose of XIFAXAN 550 mg with a single 600 mg dose of cyclosporine, an inhibitor of P-glycoprotein [see Drug Interactions (7.1)].
Cyclosporine is also an inhibitor of OATP, breast cancer resistance protein (BCRP) and a weak inhibitor of CYP3A4. The relative contribution of inhibition of each transporter by cyclosporine to the increase in rifaximin exposure is unknown.
Effect of rifaximin on other drugs
In in vitro drug interaction studies, the IC50 values for rifaximin was >50 micromolar (~60 mcg) for CYP isoforms 1A2, 2A6, 2B6, 2C9, 2C19, 2D6, and 2E1. In vitro IC50 value of rifaximin for CYP3A4 was 25 micromolar. Based on in vitro studies, clinically significant drug interaction via inhibition of 1A2, 2A6, 2B6, 2C9, 2C19, 2D6, 2E1 and 3A4 by rifaximin is not expected.
The inhibitory effect of rifaximin on P-glycoprotein transport was observed in an in vitro study. The effect of rifaximin on P-gp transporter was not evaluated in vivo.
In in vitro studies, rifaximin at 3 micromolar inhibited the uptake of estradiol glucuronide via OATP1B1 by 64% and via OATP1B3 by 70% while the uptake of estrone sulfate via OATP1A2 was inhibited by 40%. The inhibitory potential of rifaximin on these transporters at the clinically relevant concentrations is unknown.
Midazolam
In an in vitro study, rifaximin was shown to induce CYP3A4 at the concentration of 0.2 micromolar. No significant induction of CYP3A4 enzyme using midazolam as a substrate was observed when rifaximin was administered three times a day for 7 days at 200 mg and 550 mg doses in two clinical drug interaction studies in healthy subjects.
The effect of XIFAXAN 200 mg administered orally every 8 hours for 3 days and for 7 days on the pharmacokinetics of a single dose of either 2 mg intravenous midazolam or 6 mg oral midazolam was evaluated in healthy subjects. No significant difference was observed in the systemic exposure or elimination of intravenous or oral midazolam or its major metabolite, 1'-hydroxymidazolam, between midazolam alone or together with XIFAXAN. Therefore, XIFAXAN was not shown to significantly affect intestinal or hepatic CYP3A4 activity for the 200 mg three times a day dosing regimen.
When a single dose of 2 mg midazolam was orally administered after administration of XIFAXAN 550 mg three times a day for 7 days and 14 days to healthy subjects, the mean AUC of midazolam was 3.8% and 8.8% lower, respectively, than when midazolam was administered alone. The mean Cmax of midazolam was lower by 4 to 5% when XIFAXAN was administered for 7-14 days prior to midazolam administration. This degree of interaction is not considered clinically meaningful.
Oral Contraceptives Containing Ethinyl Estradiol and Norgestimate
The oral contraceptive study utilized an open-label, crossover design in 28 healthy female subjects to determine if XIFAXAN 200 mg orally administered three times a day for 3 days (the dosing regimen for travelers' diarrhea) altered the pharmacokinetics of a single dose of an oral contraceptive containing 0.07 mg ethinyl estradiol and 0.5 mg norgestimate. Results showed that the pharmacokinetics of single doses of ethinyl estradiol and norgestimate were not altered by XIFAXAN.
An open-label oral contraceptive study was conducted in 39 healthy female subjects to determine if XIFAXAN 550 mg orally administered three times a day for 7 days altered the pharmacokinetics of a single dose of an oral contraceptive containing 0.025 mg of ethinyl estradiol (EE) and 0.25 mg norgestimate (NGM). Mean Cmax of EE and NGM was lower by 25% and 13%, after the 7-day XIFAXAN regimen than when the oral contraceptive was given alone. The mean AUC values of NGM active metabolites were lower by 7% to approximately 11%, while AUC of EE was not altered in presence of rifaximin. The clinical relevance of the Cmax and AUC reductions in the presence of rifaximin is not known.
12.4. Microbiology
Mechanism of Action
Rifaximin is a semi-synthetic derivative of rifampin and acts by binding to the beta-subunit of bacterial DNA-dependent RNA polymerase blocking one of the steps in transcription. This results in inhibition of bacterial protein synthesis and consequently inhibits the growth of bacteria.
Resistance
Resistance to rifaximin is caused primarily by mutations in the rpoB gene. This changes the binding site on DNA dependent RNA polymerase and decreases rifaximin binding affinity, thereby reducing efficacy. Cross-resistance between rifaximin and other classes of antimicrobials has not been observed.
Antibacterial Activity
Rifaximin has been shown to be active against most isolates of the following microorganisms, both in vitro and in clinical infections [see Indications and Usage (1.1)]:
Aerobic bacteria
Gram-negative bacteria
Escherichia coli (enterotoxigenic and enteroaggregative strains)
Susceptibility Testing
For specific information regarding susceptibility test interpretive criteria, and associated test methods and quality control standards recognized by FDA for this drug, please see: https://www.fda.gov/STIC.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Malignant schwannomas in the heart were significantly increased in male Crl:CD (SD) rats that received rifaximin by oral gavage for two years at 150 to 250 mg/kg per day (doses equivalent to 2.4 to 4 times the recommended dose of 200 mg three times daily for TD, and equivalent to 1.3 to 2.2 times the recommended dose of 550 mg twice daily for HE, based on relative body surface area comparisons). There was no increase in tumors in Tg.rasH2 mice dosed orally with rifaximin for 26 weeks at 150 to 2000 mg/kg per day (doses equivalent to 1.2 to 16 times the recommended daily dose for TD and equivalent to 0.7 to 9 times the recommended daily dose for HE, based on relative body surface area comparisons).
Rifaximin was not genotoxic in the bacterial reverse mutation assay, chromosomal aberration assay, rat bone marrow micronucleus assay, rat hepatocyte unscheduled DNA synthesis assay, or the CHO/HGPRT mutation assay. There was no effect on fertility in male or female rats following the administration of rifaximin at doses up to 300 mg/kg (approximately 5 times the clinical dose of 600 mg per day for TD, and approximately 2.6 times the clinical dose of 1,100 mg per day for HE, adjusted for body surface area).
14. Clinical Studies
14.1 Travelers' Diarrhea
The efficacy of XIFAXAN given as 200 mg orally taken three times a day for 3 days was evaluated in 2 randomized, multi‑center, double-blind, placebo-controlled studies in adult subjects with travelers' diarrhea. One study was conducted at clinical sites in Mexico, Guatemala, and Kenya (Study 1). The other study was conducted in Mexico, Guatemala, Peru, and India (Study 2). Stool specimens were collected before treatment and 1 to 3 days following the end of treatment to identify enteric pathogens. The predominant pathogen in both studies was Escherichia coli.
The clinical efficacy of XIFAXAN was assessed by the time to return to normal, formed stools, and resolution of symptoms. The primary efficacy endpoint was time to last unformed stool (TLUS) which was defined as the time to the last unformed stool passed, after which clinical cure was declared. Table 4 displays the median TLUS and the number of patients who achieved clinical cure for the intent to treat (ITT) population of Study 1. The duration of diarrhea was significantly shorter in patients treated with XIFAXAN than in the placebo group. More patients treated with XIFAXAN were classified as clinical cures than were those in the placebo group.
Table 4. Clinical Response in Study 1 (ITT population):
| XIFAXAN (n=125) | Placebo (n=129) | Estimate (97.5% CI) | |
|---|---|---|---|
| Median TLUS (hours) | 32.5 | 58.6 | 2a (1.26, 2.50) |
| Clinical cure, n (%) | 99 (79) | 78 (60) | 19b (5.3, 32.1) |
a Hazard Ratio (p-value <0.001)
b Difference in rates (p-value <0.01)
Microbiological eradication (defined as the absence of a baseline pathogen in culture of stool after 72 hours of therapy) rates for Study 1 are presented in Table 5 for patients with any pathogen at baseline and for the subset of patients with Escherichia coli at baseline. Escherichia coli was the only pathogen with sufficient numbers to allow comparisons between treatment groups.
Even though XIFAXAN had microbiologic activity similar to placebo, it demonstrated a clinically significant reduction in duration of diarrhea and a higher clinical cure rate than placebo. Therefore, patients should be managed based on clinical response to therapy rather than microbiologic response.
Table 5. Microbiologic Eradication Rates in Study 1 Subjects with a Baseline Pathogen:
| XIFAXAN | Placebo | |
|---|---|---|
| Overall | 48/70 (69) | 41/61 (67) |
| E. coli | 38/53 (72) | 40/54 (74) |
The results of Study 2 supported the results presented for Study 1. In addition, this study provided evidence that subjects treated with XIFAXAN with fever and/or blood in the stool at baseline had prolonged TLUS. These subjects had lower clinical cure rates than those without fever or blood in the stool at baseline. Many of the patients with fever and/or blood in the stool (dysentery-like diarrheal syndromes) had invasive pathogens, primarily Campylobacter jejuni, isolated in the baseline stool.
Also in this study, the majority of the subjects treated with XIFAXAN who had Campylobacter jejuni isolated as a sole pathogen at baseline failed treatment and the resulting clinical cure rate for these patients was 23.5% (4/17). In addition to not being different from placebo, the microbiologic eradication rates for subjects with Campylobacter jejuni isolated at baseline were much lower than the eradication rates seen for Escherichia coli.
In an unrelated open-label, pharmacokinetic study of oral XIFAXAN 200 mg taken every 8 hours for 3 days, 15 adult subjects were challenged with Shigella flexneri 2a, of whom 13 developed diarrhea or dysentery and were treated with XIFAXAN. Although this open-label challenge trial was not adequate to assess the effectiveness of XIFAXAN in the treatment of shigellosis, the following observations were noted: eight subjects received rescue treatment with ciprofloxacin either because of lack of response to XIFAXAN treatment within 24 hours (2), or because they developed severe dysentery (5), or because of recurrence of Shigella flexneri 2a in the stool (1); five of the 13 subjects received ciprofloxacin although they did not have evidence of severe disease or relapse.
14.2 Hepatic Encephalopathy
The efficacy of XIFAXAN 550 mg taken orally two times a day was evaluated in a randomized, placebo-controlled, double-blind, multi-center 6-month trial of adult subjects from the U.S., Canada, and Russia who were defined as being in remission (Conn score of 0 or 1) from hepatic encephalopathy (HE). Eligible subjects had ≥2 episodes of HE associated with chronic liver disease in the previous 6 months.
A total of 299 subjects were randomized to receive either XIFAXAN (n=140) or placebo (n=159) in this study. Patients had a mean age of 56 years (range, 21-82 years), 81% <65 years of age, 61% were male and 86% White. At baseline, 67% of patients had a Conn score of 0 and 68% had an asterixis grade of 0. Patients had MELD scores of either ≤10 (27%) or 11 to 18 (64%) at baseline. No patients were enrolled with a MELD score of >25. Nine percent of the patients were Child-Pugh Class C. Lactulose was concomitantly used by 91% of the patients in each treatment arm of the study. Per the study protocol, patients were withdrawn from the study after experiencing a breakthrough HE episode. Other reasons for early study discontinuation included: adverse reactions (XIFAXAN 6%; placebo 4%), patient request to withdraw (XIFAXAN 4%; placebo 6%), and other (XIFAXAN 7%; placebo 5%).
The primary endpoint was the time to first breakthrough overt HE episode. A breakthrough overt HE episode was defined as a marked deterioration in neurological function and an increase of Conn score to Grade ≥2. In patients with a baseline Conn score of 0, a breakthrough overt HE episode was defined as an increase in Conn score of 1 and asterixis grade of 1.
Breakthrough overt HE episodes were experienced by 31 of 140 subjects (22%) in the XIFAXAN group and by 73 of 159 subjects (46%) in the placebo group during the 6-month treatment period. Comparison of Kaplan-Meier estimates of event-free curves showed XIFAXAN significantly reduced the risk of HE breakthrough by 58% during the 6-month treatment period. Presented below in Figure 1 is the Kaplan-Meier event-free curve for all subjects (n=299) in the study.
Figure 1. Kaplan-Meier Event-Free Curves1 in HE Study (Time to First Breakthroug h-HE Episode up to 6 Months of Treatment, Day 170) (ITT Population):
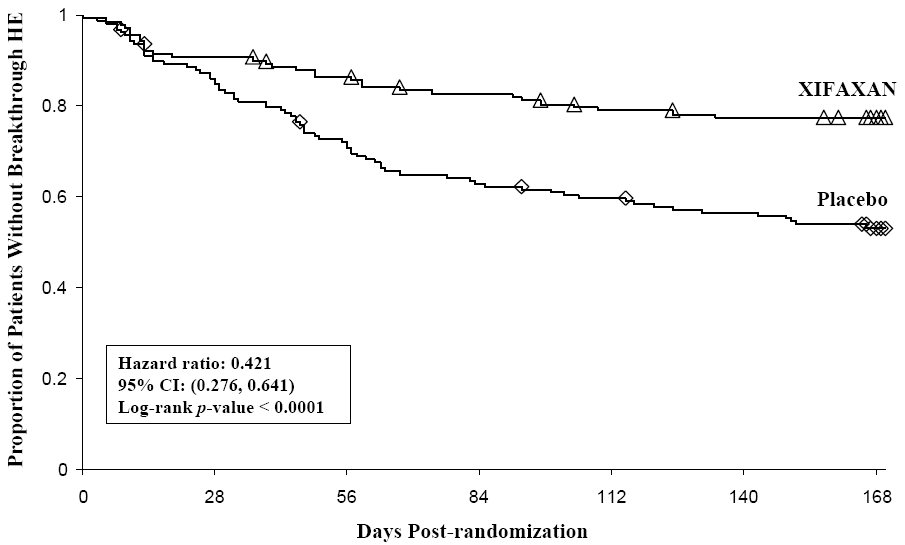
Note: Open diamonds and open triangles represent censored subjects.
1 Event-free refers to non-occurrence of breakthrough HE
When the results were evaluated by the following demographic and baseline characteristics, the treatment effect of XIFAXAN 550 mg in reducing the risk of breakthrough overt HE recurrence was consistent for: sex, baseline Conn score, duration of current remission, and diabetes. The differences in treatment effect could not be assessed in the following subpopulations due to small sample size: non-White (n=42), baseline MELD >19 (n=26), Child-Pugh Class C (n=31), and those without concomitant lactulose use (n=26).
HE-related hospitalizations (hospitalizations directly resulting from HE, or hospitalizations complicated by HE) were reported for 19 of 140 subjects (14%) and 36 of 159 subjects (23%) in the XIFAXAN (rifaximin) and placebo groups, respectively. Comparison of Kaplan-Meier estimates of event-free curves showed XIFAXAN significantly reduced the risk of HE-related hospitalizations by 50% during the 6-month treatment period. Comparison of Kaplan-Meier estimates of event-free curves is shown in Figure 2.
Figure 2. Kaplan-Meier Event-Free Curves1 in Pivotal HE Study (Time to First HE-Related Hospitalization in HE Study up to 6 Months of Treatment, Day 170) (ITT Population):
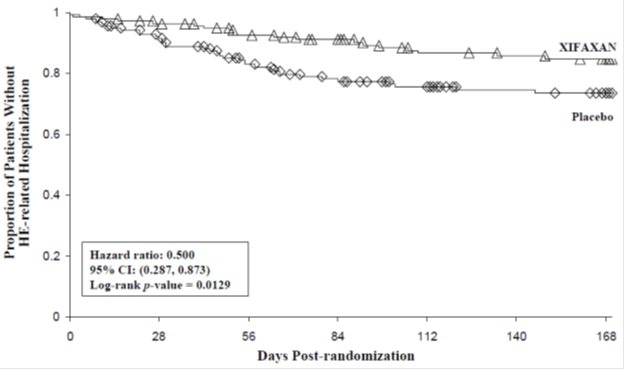
Note: Open diamonds and open triangles represent censored subjects.
1 Event-free refers to non-occurrence of HE-related hospitalization
14.3 Irritable Bowel Syndrome with Diarrhea
The efficacy of XIFAXAN for the treatment of IBS-D was established in 3 randomized, multi‑center, double-blind, placebo-controlled trials in adult patients.
Trials 1 and 2 - Design
The first two trials, Trials 1 and 2 were of identical design. In these trials, a total of 1,258 patients meeting Rome II criteria for IBS* were randomized to receive XIFAXAN 550 mg three times a day (n=624) or placebo (n=634) for 14 days and then followed for a 10-week treatment-free period. The Rome II criteria further categorizes IBS patients into 3 subtypes: diarrhea-predominant IBS (IBS-D), constipation-predominant IBS (IBS-C), or alternating IBS (bowel habits alternating between diarrhea and constipation). Patients with both IBS-D and alternating IBS were included in Trials 1 and 2. XIFAXAN is recommended for use in patients with IBS-D.
* Rome II Criteria: At least 12 weeks, which need not be consecutive, in the preceding 12 months of abdominal discomfort or pain that has two out of three features: 1. Relieved with defecation; and/or 2. Onset associated with a change in frequency of stool; and/or 3. Onset associated with a change in form (appearance) of stool.
Symptoms that Cumulatively Support the Diagnosis of Irritable Bowel Syndrome:
- Abnormal stool frequency (for research purposes "abnormal" may be defined as greater than 3 bowel movements per day and less than 3 bowel movements per week); Abnormal stool form (lumpy/hard or loose/watery stool); Abnormal stool passage (straining, urgency, or feeling of incomplete evacuation); Passage of mucus; Bloating or feeling of abdominal distension.
Trial 3 - Design
Trial 3 evaluated repeat treatment in adults with IBS-D meeting Rome III criteria** for up to 46 weeks. A total of 2,579 patients were enrolled to receive open-label XIFAXAN for 14 days. Of 2,438 evaluable patients, 1,074 (44%) responded to initial treatment and were evaluated over 22 weeks for continued response or recurrence of IBS-symptoms. A total of 636 patients had symptom recurrence and were randomized into the double-blind phase of the study. These patients were scheduled to receive XIFAXAN 550 mg three times a day (n=328) or placebo (n=308) for two additional 14-day repeat treatment courses separated by 10 weeks. See Figure 3.
Figure 3. Trial 3 Study Design:
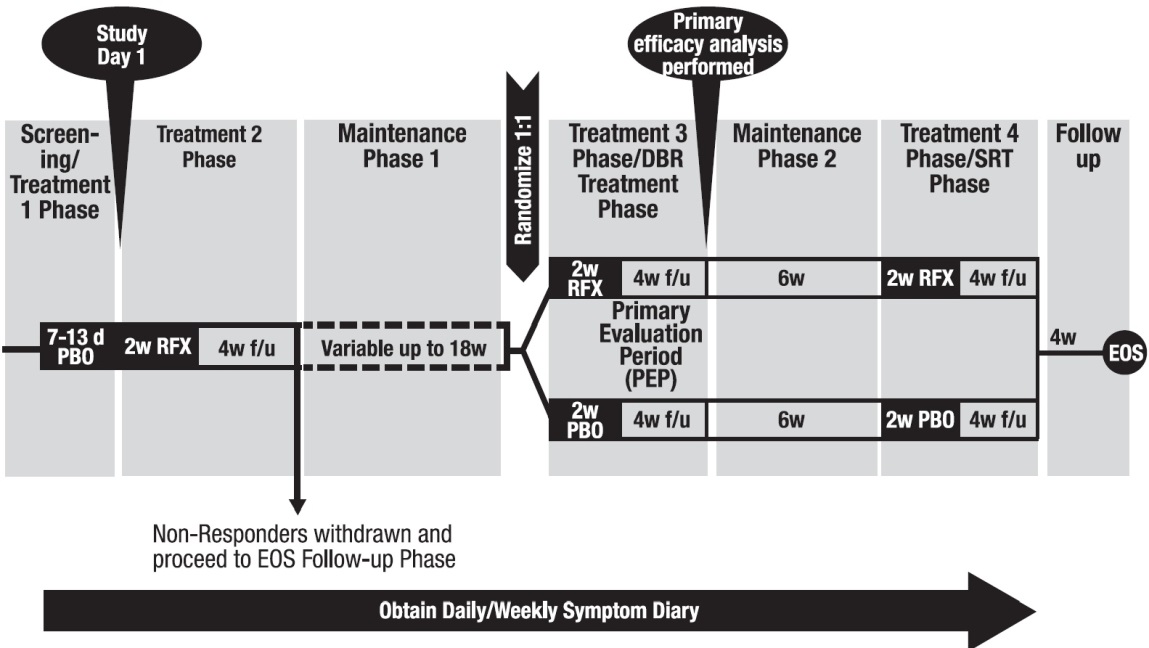
The IBS-D population from the three studies had mean age of 47 (range: 18 to 88) years of which approximately 11% of patients were ≥65 years old, 72% were female, and 88% were White.
** Rome III Criteria: Recurrent abdominal pain or discomfort (uncomfortable sensation not described as pain) at least 3 days/month in last 3 months associated with two or more of the following: 1. Improvement with defecation; 2. Onset associated with a change in frequency of stool; 3. Onset associated with a change in form (appearance) of stool.
Trials 1 and 2 - Results
Trials 1 and 2 included 1,258 IBS-D patients (309 XIFAXAN, 314 placebo); (315 XIFAXAN, 320 placebo). The primary endpoint for both trials was the proportion of patients who achieved adequate relief of IBS signs and symptoms for at least 2 of 4 weeks during the month following 14 days of treatment. Adequate relief was defined as a response of "yes" to the following weekly Subject Global Assessment (SGA) question: "In regards to your IBS symptoms, compared to the way you felt before you started study medication, have you, in the past 7 days, had adequate relief of your IBS symptoms? [Yes/No]."
Adequate relief of IBS symptoms was experienced by more patients receiving XIFAXAN than those receiving placebo during the month following 2 weeks of treatment (SGA-IBS Weekly Results: 41% vs. 31%, p=0.0125; 41% vs. 32%, p=0.0263) (See Table 6).
Table 6. Adequate Relief of IBS Symptoms During the Month Following Two Weeks of Treatment:
| Endpoint | Trial 1 | ||
|---|---|---|---|
| XIFAXAN n=309 n (%) | Placebo n=314 n (%) | Treatment Difference (95% CIa) | |
| Adequate Relief of IBS Symptomsb | 126 (41) | 98 (31) | 10% (2.1%, 17.1%) |
| Endpoint | Trial 2 | ||
| XIFAXAN n=315 n (%) | Placebo n=320 n (%) | Treatment Difference (95% CIa) | |
| Adequate Relief of IBS Symptomsb | 128 (41) | 103 (32) | 8% (1.0%, 15.9%) |
a Confidence Interval
b The p-value for the primary endpoint for Trial 1 and for Trial 2 was <0.05
The trials examined a composite endpoint which defined responders by IBS-related abdominal pain and stool consistency measures. Patients were monthly responders if they met both of the following criteria:
- experienced a ≥30% decrease from baseline in abdominal pain for ≥2 weeks during the month following 2 weeks of treatment
- had a weekly mean stool consistency score <4 (loose stool) for ≥2 weeks during the month following 2 weeks of treatment
More patients receiving XIFAXAN were monthly responders for abdominal pain and stool consistency in Trials 1 and 2 (see Table 7).
Table 7. Efficacy Responder Rates in Trial 1 and 2 During the Month Following Two Weeks of Treatment:
| Endpoint | Trial 1 | ||
|---|---|---|---|
| XIFAXAN n=309 n (%) | Placebo n=314 n (%) | Treatment Difference (95% CIª) | |
| Abdominal Pain and Stool Consistency Responders b | 144 (47) | 121 (39) | 8% (0.3%, 15.9%) |
| Abdominal Pain Responders | 159 (51) | 132 (42) | 9% (1.8%, 17.5%) |
| Stool Consistency Responders | 244 (79) | 212 (68) | 11% (4.4%, 18.2%) |
| Endpoint | Trial 2 | ||
|---|---|---|---|
| XIFAXAN n=315 n (%) | Placebo n=320 n (%) | Treatment Difference (95% CIa) | |
| Abdominal Pain and Stool Consistency Respondersb | 147 (47) | 116 (36) | 11% (2.7%, 18.0%) |
| Abdominal Pain Responders | 165 (52) | 138 (43) | 9% (1.5%, 17.0%) |
| Stool Consistency Responders | 233 (74) | 206 (64) | 10% (2.3%, 16.7%) |
a Confidence Interval
b The p-value for the composite endpoint for Trial 1 and 2 was <0.05 and <0.01, respectively
Trial 3 - Results
In TARGET 3, 2,579 patients were scheduled to receive an initial 14-day course of open-label XIFAXAN followed by 4 weeks of treatment-free follow-up. At the end of the follow-up period, patients were assessed for response to treatment. Patients were considered a responder if they achieved both of the following:
- ≥30% improvement from baseline in the weekly average abdominal pain score based on the daily question: "In regards to your specific IBS symptoms of abdominal pain, on a scale of 0-10, what was your worst IBS-related abdominal pain over the last 24 hours? 'Zero' means you have no pain at all; 'Ten' means the worst possible pain you can imagine".
- at least a 50% reduction in the number of days in a week with a daily stool consistency of Bristol Stool Scale type 6 or 7 compared with baseline where 6=fluffy pieces with ragged edges, a mushy stool; 7=watery stool, no solid pieces; entirely liquid.
Responders were then followed for recurrence of their IBS-related symptoms of abdominal pain or mushy/watery stool consistency for up to 20 treatment-free weeks.
When patients experienced recurrence of their symptoms of abdominal pain or mushy/watery stool consistency for 3 weeks of a rolling 4-week period, they were randomized into the double-blind, placebo-controlled repeat treatment phase. Of 1,074 patients who responded to open-label XIFAXAN, 382 experienced a period of symptom inactivity or decrease that did not require repeat treatment by the time they discontinued, including patients who completed the 22 weeks after initial treatment with XIFAXAN. See Figure 3.
Overall, 1,257 of 2,579 patients (49%) were nonresponders in the open-label phase and per the study protocol were withdrawn from the study. Other reasons for discontinuation include: patient request (5%), patient lost to follow-up (4%), adverse reaction (3%), and other (0.8%).
There were 1,074 (44%) of 2,438 evaluable patients who responded to initial treatment with improvement in abdominal pain and stool consistency. The response rate for each IBS symptom during the open-label phase of Trial 3 is similar to the rates seen in Trials 1 and 2 (see Table 7). A total of 636 patients subsequently had sign and symptom recurrence and were randomized to the repeat treatment phase. The median time to recurrence for patients who experienced initial response during the open-label phase with XIFAXAN was 10 weeks (range 6 to 24 weeks).
The XIFAXAN (rifaximin) and placebo treatment groups had similar baseline IBS symptom scores at the time of recurrence and randomization to the double-blind phase, but symptom scores were less severe than at study entry into the open-label phase.
Patients were deemed to have recurrent signs and symptoms by the following criteria: a return of abdominal pain or lack of stool consistency for at least 3 weeks during a 4-week follow-up period. The primary endpoint in the double-blind, placebo-controlled portion of the trial was the proportion of patients who were responders to repeat treatment in both IBS-related abdominal pain and stool consistency as defined above during the 4 weeks following the first repeat treatment with XIFAXAN. The primary analysis was performed using the worst case analysis method where patients with <4 days of diary entries in a given week are considered as non-responders for that week.
More patients receiving XIFAXAN were monthly responders for abdominal pain and stool consistency in the primary analysis in Trial 3 (see Table 8).
Table 8. Efficacy Responder Rates in Trial 3 in a Given Week for at Least 2 Weeks During Weeks 3 to 6 of the Double-Blind, First Repeat Treatment Phase:
| Placebo (n=308) n (%) | XIFAXAN (n=328) n (%) | Treatment Difference (95% CIa) | |
|---|---|---|---|
| Combined Responderb: Abdominal Pain and Stool Consistency Respondersc | 97 (31) | 125 (38) | 7% (0.9%, 16.9%) |
| Abdominal Pain Responders (≥30% reduction in abdominal pain) | 130 (42) | 166 (51) | 9% (1.6%, 17.0%) |
| Stool Consistency Responders (≥50% reduction from baseline in days/week with loose or watery stools) | 154 (50) | 170 (52) | 2% (-4.7%, 11.0%) |
a Confidence Intervals were derived based on CMH test adjusting for center and patients' time to recurrence during maintenance phase
b Primary endpoint
c Subjects were IBS-related abdominal pain and stool consistency responders if they were both weekly IBS-related abdominal pain responders and weekly stool consistency responders in a given week for at least 2 weeks during Weeks 3 to 6 in the double-blind first repeat treatment phase. Weekly responder in IBS-related abdominal pain was defined as a 30% or greater improvement from baseline in the weekly average abdominal pain score. Weekly responder in stool consistency was defined as a 50% or greater reduction in the number of days in a week with stool consistency of type 6 or 7 compared with baseline. The p-value for this composite endpoint was <0.05
Thirty-six of 308 (11.7%) of placebo patients and 56 of 328 (17.1%) of XIFAXAN-treated patients responded to the first repeat treatment and did not have recurrence of signs and symptoms through the treatment-free follow-up period (10 weeks after first repeat treatment). The response rate difference was 5.4% with 95% confidence interval (1.2% to 11.6%).
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.