XOLAIR 150 mg Solution for injection Ref.[8001] Active ingredients: Omalizumab
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: Novartis Europharm Limited, Vista Building, Elm Park, Merrion Road, Dublin 4, Ireland
Pharmacodynamic properties
Pharmacotherapeutic group: Drugs for obstructive airway diseases, other systemic drugs for obstructive airway diseases
ATC code: R03DX05
Allergic asthma and chronic rhinosinusitis with nasal polyps (CRSwNP)
Mechanism of action
Omalizumab is a recombinant DNA-derived humanised monoclonal antibody that selectively binds to human immunoglobulin E (IgE) and prevents binding of IgE to FcεRI (high-affinity IgE receptor) on basophils and mast cells, thereby reducing the amount of free IgE that is available to trigger the allergic cascade. The antibody is an IgG1 kappa that contains human framework regions with the complementary-determining regions of a murine parent antibody that binds to IgE.
Treatment of atopic subjects with omalizumab resulted in a marked down-regulation of FcεRI receptors on basophils. Omalizumab inhibits IgE-mediated inflammation, as evidenced by reduced blood and tissue eosinophils and reduced inflammatory mediators, including IL-4, IL-5, and IL-13 by innate, adaptive and non-immune cells.
Pharmacodynamic effects
Allergic asthma
The in vitro histamine release from basophils isolated from omalizumab-treated subjects was reduced by approximately 90% following stimulation with an allergen compared to pre-treatment values.
In clinical studies in allergic asthma patients, serum free IgE levels were reduced in a dose-dependent manner within one hour following the first dose and maintained between doses. One year after discontinuation of omalizumab dosing, the IgE levels had returned to pre-treatment levels with no observed rebound in IgE levels after washout of the medicinal product.
Chronic rhinosinusitis with nasal polyps (CRSwNP)
In clinical studies in patients with CRSwNP, omalizumab treatment led to a reduction in serum free IgE (approx. 95%) and an increase in serum total IgE levels, to a similar extent as observed in patients with allergic asthma. Total IgE levels in serum increased due to the formation of omalizumab-IgE complexes that have a slower elimination rate compared with free IgE.
Chronic spontaneous urticaria (CSU)
Mechanism of action
Omalizumab is a recombinant DNA-derived humanised monoclonal antibody that selectively binds to human immunoglobulin E (IgE) and lowers free IgE levels. The antibody is an IgG1 kappa that contains human framework regions with the complementary-determining regions of a murine parent antibody that binds to IgE. Subsequently, IgE receptors (FcεRI) on cells down-regulate. It is not entirely understood how this results in an improvement of CSU symptoms.
Pharmacodynamic effects
In clinical studies in CSU patients, maximum suppression of free IgE was observed 3 days after the first subcutaneous dose. After repeated dosing once every 4 weeks, pre-dose serum free IgE levels remained stable between 12 and 24 weeks of treatment. After discontinuation of omalizumab, free IgE levels increased towards pre-treatment levels over a 16-week treatment-free follow-up period.
Clinical efficacy and safety
Allergic asthma
Adults and adolescents ≥12 years of age
The efficacy and safety of omalizumab were demonstrated in a 28-week double-blind placebo- controlled study (study 1) involving 419 severe allergic asthmatics, ages 12-79 years, who had reduced lung function (FEV1 40-80% predicted) and poor asthma symptom control despite receiving high dose inhaled corticosteroids and a long-acting beta2-agonist. Eligible patients had experienced multiple asthma exacerbations requiring systemic corticosteroid treatment or had been hospitalised or attended an emergency room due to a severe asthma exacerbation in the past year despite continuous treatment with high-dose inhaled corticosteroids and a long-acting beta2-agonist. Subcutaneous omalizumab or placebo were administered as add-on therapy to >1 000 micrograms beclomethasone dipropionate (or equivalent) plus a long-acting beta2-agonist. Oral corticosteroid, theophylline and leukotriene-modifier maintenance therapies were allowed (22%, 27%, and 35% of patients, respectively).
The rate of asthma exacerbations requiring treatment with bursts of systemic corticosteroids was the primary endpoint. Omalizumab reduced the rate of asthma exacerbations by 19% (p=0.153). Further evaluations which did show statistical significance (p<0.05) in favour of omalizumab included reductions in severe exacerbations (where patient's lung function was reduced to below 60% of personal best and requiring systemic corticosteroids) and asthma-related emergency visits (comprised of hospitalisations, emergency room, and unscheduled doctor visits), and improvements in Physician's overall assessment of treatment effectiveness, Asthma-related Quality of Life (AQL), asthma symptoms and lung function.
In a subgroup analysis, patients with pre-treatment total IgE ≥76 IU/ml were more likely to experience clinically meaningful benefit to omalizumab. In these patients in study 1 omalizumab reduced the rate of asthma exacerbations by 40% (p=0.002). In addition more patients had clinically meaningful responses in the total IgE ≥76 IU/ml population across the omalizumab severe asthma programme. Table 6 includes results in the study 1 population.
Table 6. Results of study 1:
| Whole study 1 population | ||
|---|---|---|
| Omalizumab N=209 | Placebo N=210 | |
| Asthma exacerbations | ||
| Rate per 28-week period | 0.74 | 0.92 |
| % reduction, p-value for rate ratio | 19.4%, p=0.153 | |
| Severe asthma exacerbations | ||
| Rate per 28-week period | 0.24 | 0.48 |
| % reduction, p-value for rate ratio | 50.1%, p=0.002 | |
| Emergency visits | ||
| Rate per 28-week period | 0.24 | 0.43 |
| % reduction, p-value for rate ratio | 43.9%, p=0.038 | |
| Physician's overall assessment | ||
| % responders* | 60.5% | 42.8% |
| p-value** | <0.001 | |
| AQL improvement | ||
| % of patients ≥0.5 improvement | 60.8% | 47.8% |
| p-value | 0.008 | |
* marked improvement or complete control
** p-value for overall distribution of assessment
Study 2 assessed the efficacy and safety of omalizumab in a population of 312 severe allergic asthmatics which matched the population in study 1. Treatment with omalizumab in this open label study led to a 61% reduction in clinically significant asthma exacerbation rate compared to current asthma therapy alone.
Four additional large placebo-controlled supportive studies of 28 to 52 weeks duration in 1 722 adults and adolescents (studies 3, 4, 5, 6) assessed the efficacy and safety of omalizumab in patients with severe persistent asthma. Most patients were inadequately controlled but were receiving less concomitant asthma therapy than patients in studies 1 or 2. Studies 3-5 used exacerbation as primary endpoint, whereas study 6 primarily evaluated inhaled corticosteroid sparing.
In studies 3, 4 and 5 patients treated with omalizumab had respective reductions in asthma exacerbation rates of 37.5% (p=0.027), 40.3% (p<0.001) and 57.6% (p<0.001) compared to placebo.
In study 6, significantly more severe allergic asthma patients on omalizumab were able to reduce their fluticasone dose to ≤500 micrograms/day without deterioration of asthma control (60.3%) compared to the placebo group (45.8%, p<0.05).
Quality of life scores were measured using the Juniper Asthma-related Quality of Life Questionnaire. For all six studies there was a statistically significant improvement from baseline in quality of life scores for omalizumab patients versus the placebo or control group.
Physician's overall assessment of treatment effectiveness: Physician's overall assessment was performed in five of the above studies as a broad measure of asthma control performed by the treating physician. The physician was able to take into account PEF (peak expiratory flow), day and night time symptoms, rescue medicinal product use, spirometry and exacerbations. In all five studies a significantly greater proportion of omalizumab-treated patients were judged to have achieved either a marked improvement or complete control of their asthma compared to placebo patients.
Children 6 to <12 years of age
The primary support for safety and efficacy of omalizumab in the group aged 6 to <12 years comes from one randomised, double-blind, placebo-controlled, multi-centre trial (study 7).
Study 7 was a placebo-controlled trial which included a specific subgroup (n=235) of patients as defined in the present indication, who were treated with high-dose inhaled corticosteroids (≥500 μg/day fluticasone equivalent) plus long-acting beta agonist.
A clinically significant exacerbation was defined as a worsening of asthma symptoms as judged clinically by the investigator, requiring doubling of the baseline inhaled corticosteroid dose for at least 3 days and/or treatment with rescue systemic (oral or intravenous) corticosteroids for at least 3 days.
In the specific subgroup of patients on high dose inhaled corticosteroids, the omalizumab group had a statistically significantly lower rate of clinically significant asthma exacerbations than the placebo group. At 24 weeks, the difference in rates between treatment groups represented a 34% (rate ratio 0.662, p=0.047) decrease relative to placebo for omalizumab patients. In the second double-blind 28-week treatment period the difference in rates between treatment groups represented a 63% (rate ratio 0.37, p<0.001) decrease relative to placebo for omalizumab patients.
During the 52-week double-blind treatment period (including the 24-week fixed-dose steroid phase and the 28-week steroid adjustment phase) the difference in rates between treatment groups represented a 50% (rate ratio 0.504, p<0.001) relative decrease in exacerbations for omalizumab patients.
The omalizumab group showed greater decreases in beta-agonist rescue medicinal product use than the placebo group at the end of the 52-week treatment period, although the difference between treatment groups was not statistically significant. For the global evaluation of treatment effectiveness at the end of the 52-week double-blind treatment period in the subgroup of severe patients on high- dose inhaled corticosteroids plus long-acting beta agonists, the proportion of patients rated as having 'excellent' treatment effectiveness was higher, and the proportions having 'moderate' or 'poor' treatment effectiveness lower in the omalizumab group compared to the placebo group; the difference between groups was statistically significant (p<0.001), while there were no differences between the omalizumab and placebo groups for patients' subjective Quality of Life ratings.
Chronic rhinosinusitis with nasal polyps (CRSwNP)
The safety and efficacy of omalizumab were evaluated in two randomised, double-blind, placebo- controlled trials in patients with CRSwNP (Table 7). Patients received omalizumab or placebo subcutaneously every 2 or 4 weeks (see section 4.2). All patients received background intranasal mometasone therapy throughout the study. Prior sino-nasal surgery or prior systemic corticosteroid usage were not required for inclusion in the studies. Patients received omalizumab or placebo for 24 weeks followed by a 4-week follow-up period. Demographics and baseline characteristics, including allergic comorbidities, are described in Table 7.
Table 7. Demographics and baseline characteristics of nasal polyp studies:
| Parameter | Nasal polyp study 1 N=138 | Nasal polyp study 2 N=127 |
|---|---|---|
| Mean age (years) (SD) | 51.0 (13.2) | 50.1 (11.9) |
| % Male | 63.8 | 65.4 |
| Patients with systemic corticosteroid use in the previous year (%) | 18.8 | 26.0 |
| Bilateral endoscopic nasal polyp score (NPS): mean (SD), range 0-8 | 6.2 (1.0) | 6.3 (0.9) |
| Nasal congestion score (NCS): mean (SD), range 0-3 | 2.4 (0.6) | 2.3 (0.7) |
| Sense of smell score: mean (SD), range 0-3 | 2.7 (0.7) | 2.7 (0.7) |
| SNOT-22 total score: mean (SD) range 0-110 | 60.1 (17.7) | 59.5 (19.3) |
| Blood eosinophils (cells/μl): mean (SD) | 346.1 (284.1) | 334.6 (187.6) |
| Total IgE IU/ml: mean (SD) | 160.9 (139.6) | 190.2 (200.5) |
| Asthma (%) | 53.6 | 60.6 |
| Mild (%) | 37.8 | 32.5 |
| Moderate (%) | 58.1 | 58.4 |
| Severe (%) | 4.1 | 9.1 |
| Aspirin exacerbated respiratory disease (%) | 19.6 | 35.4 |
| Allergic rhinitis | 43.5 | 42.5 |
SD = standard deviation; SNOT-22 = Sino-Nasal Outcome Test 22 Questionnaire; IgE = Immunoglobulin E; IU = international units. For NPS, NCS, and SNOT-22 higher scores indicate greater disease severity.
The co-primary endpoints were bilateral nasal polyps score (NPS) and average daily nasal congestion score (NCS) at Week 24. In both nasal polyp studies 1 and 2, patients who received omalizumab had statistically significant greater improvements from baseline at Week 24 in NPS and weekly average NCS than patients who received placebo. Results from nasal polyp studies 1 and 2 are shown in Table 8.
Table 8. Change from baseline at Week 24 in clinical scores from nasal polyp study 1, nasal polyp study 2, and pooled data:
| Nasal polyp study 1 | Nasal polyp study 2 | Nasal polyp pooled results | ||||
|---|---|---|---|---|---|---|
| Placebo | Omalizumab | Placebo | Omalizumab | Placebo | Omalizumab | |
| N | 66 | 72 | 65 | 62 | 131 | 134 |
| Nasal polyp score | ||||||
| Baseline mean | 6.32 | 6.19 | 6.09 | 6.44 | 6.21 | 6.31 |
| LS mean change at Week 24 | 0.06 | -1.08 | -0.31 | -0.90 | -0.13 | -0.99 |
| Difference (95% CI) | -1.14 (-1.59, -069) | -0.59 (-1.05, -012) | -0.86 (-1.18, -0.54) | |||
| p-value | <0.0001 | 0.0140 | <0.0001 | |||
| 7-day average of daily nasal congestion score | ||||||
| Baseline mean | 2.46 | 2.40 | 2.29 | 2.26 | 2.38 | 2.34 |
| LS mean change at Week 24 | -0.35 | -0.89 | -0.20 | -0.70 | -0.28 | -0.80 |
| Difference (95% CI) | -0.55 (-0.84, -0.25) | -0.50 (-0.80, -0.19) | -0.52 (-0.73, -0.31) | |||
| p-value | 0.0004 | 0.0017 | <0.0001 | |||
| TNSS | ||||||
| Baseline mean | 9.33 | 8.56 | 8.73 | 8.37 | 9.03 | 8.47 |
| LS mean change at Week 24 | -1.06 | -2.97 | -0.44 | -2.53 | -0.77 | -2.75 |
| Difference (95% CI) | -1.91 (-2.85, -0.96) | -2.09 (-3.00, -1.18) | -1.98 (-2.63, -1.33) | |||
| p-value | 0.0001 | <0.0001 | <0.0001 | |||
| SNOT-22 | ||||||
| Baseline mean | 60.26 | 59.82 | 59.80 | 59.21 | 60.03 | 59.54 |
| LS mean change at Week 24 | -8.58 | -24.70 | -6.55 | -21.59 | -7.73 | -23.10 |
| Difference (95% CI) | -16.12 (-21.86, -10.38) | -15.04 (-21.26, -8.82) | -15.36 (-19.57, -11.16) | |||
| p-value (MID = 8.9) | <0.0001 | <0.0001 | <0.0001 | |||
| UPSIT | ||||||
| Baseline mean | 13.56 | 12.78 | 13.27 | 12.87 | 13.41 | 12.82 |
| LS mean change at Week 24 | 0.63 | 4.44 | 0.44 | 4.31 | 0.54 | 4.38 |
| Difference (95% CI) | 3.81 (1.38, 6.24) | 3.86 (1.57, 6.15) | 3.84 (2.17, 5.51) | |||
| p-value | 0.0024 | 0.0011 | <0.0001 | |||
LS = least-square; CI = confidence interval; TNSS = Total nasal symptom score; SNOT-22 = Sino-Nasal Outcome Test 22 Questionnaire; UPSIT = University of Pennsylvania Smell Identification Test; MID = minimal important difference.
Figure 1. Mean change from baseline in nasal congestion score and mean change from baseline in nasal polyp score by treatment group in nasal polyp study 1 and study 2:
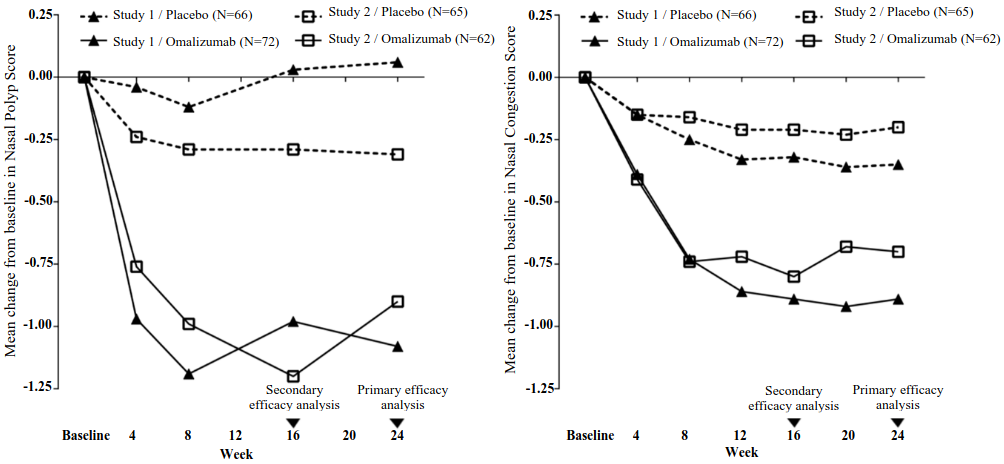
In a pre-specified pooled analysis of rescue treatment (systemic corticosteroids for ≥3 consecutive days or nasal polypectomy) during the 24-week treatment period, the proportion of patients requiring rescue treatment was lower in omalizumab compared to placebo (2.3% versus 6.2%, respectively). The odds-ratio of having taken rescue treatment in omalizumab compared to placebo was 0.38 (95% CI: 0.10, 1.49). There were no sino-nasal surgeries reported in either study.
The long-term efficacy and safety of omalizumab in patients with CRSwNP who had participated in nasal polyp studies 1 and 2 was assessed in an open-label extension study. Efficacy data from this study suggest that clinical benefit provided at Week 24 was sustained through to Week 52. Safety data were overall consistent with the known safety profile of omalizumab.
Chronic spontaneous urticaria (CSU)
The efficacy and safety of omalizumab were demonstrated in two randomised, placebo-controlled phase III studies (study 1 and 2) in patients with CSU who remained symptomatic despite H1 antihistamine therapy at the approved dose. A third study (study 3) primarily evaluated the safety of omalizumab in patients with CSU who remained symptomatic despite treatment with H1 antihistamines at up to four times the approved dose and H2 antihistamine and/or LTRA treatment. The three studies enrolled 975 patients aged between 12 and 75 years (mean age 42.3 years; 39 patients 12-17 years, 54 patients ≥65 years; 259 males and 716 females). All patients were required to have inadequate symptom control, as assessed by a weekly urticaria activity score (UAS7, range 0-42) of ≥16, and a weekly itch severity score (which is a component of the UAS7; range 0-21) of ≥8 for the 7 days prior to randomisation, despite having used an antihistamine for at least 2 weeks beforehand.
In studies 1 and 2, patients had a mean weekly itch severity score of between 13.7 and 14.5 at baseline and a mean UAS7 score of 29.5 and 31.7 respectively. Patients in safety study 3 had a mean weekly itch severity score of 13.8 and a mean UAS7 score of 31.2 at baseline. Across all three studies, patients reported receiving on average 4 to 6 medicinal products (including H1 antihistamines) for CSU symptoms prior to study enrollment. Patients received omalizumab at 75 mg, 150 mg or 300 mg or placebo by subcutaneous injection every 4 weeks for 24 and 12 weeks in studies 1 and 2, respectively, and 300 mg or placebo by subcutaneous injection every 4 weeks for 24 weeks in study 3. All studies had a 16-week treatment-free follow-up period.
The primary endpoint was the change from baseline to week 12 in weekly itch severity score. Omalizumab at 300 mg reduced the weekly itch severity score by 8.55 to 9.77 (p<0.0001) compared to a reduction of 3.63 to 5.14 for placebo (see Table 9). Statistically significant results were further observed in the responder rates for UAS7≤6 (at week 12) which were higher for the 300 mg treatment groups, ranging from 52-66% (p<0.0001) compared to 11-19% for the placebo groups, and complete response (UAS7=0) was achieved by 34-44% (p<0.0001) of patients treated with 300 mg compared to 5-9% of patients in the placebo groups. Patients in the 300 mg treatment groups achieved the highest mean proportion of angioedema-free days from week 4 to week 12, (91.0-96.1%; p<0.001) compared to the placebo groups (88.1-89.2%). Mean change from baseline to week 12 in the overall DLQI for the 300 mg treatment groups was greater (p<0.001) than for placebo showing an improvement ranging from 9.7-10.3 points compared to 5.1-6.1 points for the corresponding placebo groups.
Table 9. Change from baseline to week 12 in weekly itch severity score, studies 1, 2 and 3 (mITT population*):
| Placebo | Omalizumab 300 mg | |
|---|---|---|
| Study 1 | ||
| N | 80 | 81 |
| Mean (SD) | −3.63 (5.22) | −9.40 (5.73) |
| Difference in LS means vs. placebo1 | - | −5.80 |
| 95% CI for difference | - | −7.49,−4.10 |
| P-value vs. placebo2 | - | <0.0001 |
| Study 2 | ||
| N | 79 | 79 |
| Mean (SD) | −5.14 (5.58) | −9.77 (5.95) |
| Difference in LS means vs. placebo1 | - | −4.81 |
| 95% CI for difference | - | −6.49,−3.13 |
| P-value vs. placebo2 | - | <0.0001 |
| Study 3 | ||
| N | 83 | 252 |
| Mean (SD) | −4.01 (5.87) | −8.55 (6.01) |
| Difference in LS means vs. placebo1 | - | -4.52 |
| 95% CI for difference | - | −5.97, −3.08 |
| P-value vs. placebo2 | - | <0.0001 |
* Modified intent-to-treat (mITT) population: included all patients who were randomised and received at least one dose of study medicinal product.
BOCF (Baseline Observation Carried Forward) was used to impute missing data.
1 The LS mean was estimated using an ANCOVA model. The strata were baseline weekly itch severity score (<13 vs. ≥13) and baseline weight (<80 kg vs. ≥80 kg).
2 p-value is derived from ANCOVA t-test.
Figure 2 shows the mean weekly itch severity score over time in study 1. The mean weekly itch severity scores significantly decreased with a maximum effect around week 12 that was sustained over the 24-week treatment period. The results were similar in study 3.
In all three studies the mean weekly itch severity score increased gradually during the 16-week treatment-free follow-up period, consistent with symptom re-occurrence. Mean values at the end of the follow-up period were similar to the placebo group, but lower than respective mean baseline values.
Figure 2. Mean weekly itch severity score over time, study 1 (mITT population):
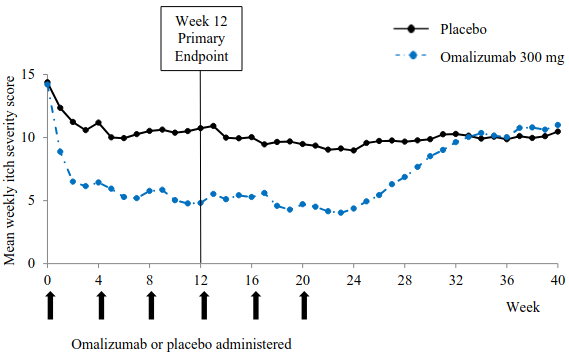
BOCF = baseline observation carried forward; mITT = modified intention-to-treat population
The magnitude of the efficacy outcomes observed at week 24 of treatment was comparable to that observed at week 12: For 300 mg, in studies 1 and 3, the mean decrease from baseline in weekly itch severity score was 9.8 and 8.6, the proportion of patients with UAS7≤6 was 61.7% and 55.6%, and the proportion of patients with complete response (UAS7=0) was 48.1% and 42.5%, respectively, (all p<0.0001, when compared to placebo).
Clinical trial data on adolescents (12 to 17 years) included a total of 39 patients, of whom 11 received the 300 mg dose. Results for the 300 mg are available for 9 patients at week 12 and 6 patients at week 24, and show a similar magnitude of response to omalizumab treatment compared to the adult population. Mean change from baseline in weekly itch severity score showed a reduction of 8.25 at week 12 and of 8.95 at week 24. The responder rates were: 33% at week 12 and 67% at week 24 for UAS7=0, and 56% at week 12 and 67% at week 24 for UAS7≤6.
In a 48-week study, 206 patients aged between 12 and 75 years were enrolled into a 24-week open-label treatment period of omalizumab 300 mg every 4 weeks. Patients who responded to treatment in this open-label period were then randomised to receive omalizumab 300 mg (81 patients) or placebo (53 patients) every 4 weeks for an additional 24 weeks.
Of the patients who remained on omalizumab treatment for 48 weeks, 21% experienced clinical worsening (UAS7 score ≥12 for at least 2 consecutive weeks post-randomisation between weeks 24 and 48), versus 60.4% of those treated with placebo at week 48 (difference ˗39.4%, p<0.0001, 95% CI: −54.5%, −22.5%).
Pharmacokinetic properties
The pharmacokinetics of omalizumab have been studied in adult and adolescent patients with allergic asthma as well as in adult patients with CRSwNP, and adult and adolescent patients with CSU. The general pharmacokinetic characteristics of omalizumab are similar in these patient populations.
Absorption
After subcutaneous administration, omalizumab is absorbed with an average absolute bioavailability of 62%. Following a single subcutaneous dose in adult and adolescent patients with asthma or CSU, omalizumab was absorbed slowly, reaching peak serum concentrations after an average of 6-8 days. In patients with asthma, following multiple doses of omalizumab, areas under the serum concentration- time curve from Day 0 to Day 14 at steady state were up to 6-fold of those after the first dose.
The pharmacokinetics of omalizumab are linear at doses greater than 0.5 mg/kg. Following doses of 75 mg, 150 mg or 300 mg every 4 weeks in patients with CSU, trough serum concentrations of omalizumab increased proportionally with the dose level.
Administration of Xolair manufactured as a lyophilised or liquid formulation resulted in similar serum concentration-time profiles of omalizumab.
Distribution
In vitro, omalizumab forms complexes of limited size with IgE. Precipitating complexes and complexes larger than one million Daltons in molecular weight are not observed in vitro or in vivo. Based on population pharmacokinetics, distribution of omalizumab was similar in patients with allergic asthma and patients with CSU. The apparent volume of distribution in patients with asthma following subcutaneous administration was 78 ± 32 ml/kg.
Elimination
Clearance of omalizumab involves IgG clearance processes as well as clearance via specific binding and complex formation with its target ligand, IgE. Liver elimination of IgG includes degradation in the reticuloendothelial system and endothelial cells. Intact IgG is also excreted in bile. In asthma patients the omalizumab serum elimination half-life averaged 26 days, with apparent clearance averaging 2.4 ± 1.1 ml/kg/day. Doubling of body weight approximately doubled apparent clearance. In CSU patients, based on population pharmacokinetic simulations, omalizumab serum elimination half-life at steady state averaged 24 days and apparent clearance at steady state for a patient of 80 kg weight was 3.0 ml/kg/day.
Characteristics in patient populations
Age, Race/Ethnicity, Gender, Body Mass Index
Patients with allergic asthma and chronic rhinosinusitis with nasal polyps (CRSwNP):
The population pharmacokinetics of omalizumab were analysed to evaluate the effects of demographic characteristics. Analyses of these limited data suggest that no dose adjustments are necessary for age (6-76 years for patients with allergic asthma; 18 to 75 for patients with CRSwNP), race/ethnicity, gender or body mass index (see section 4.2).
Patients with CSU:
The effects of demographic characteristics and other factors on omalizumab exposure were evaluated based on population pharmacokinetics. In addition, covariate effects were evaluated by analysing the relationship between omalizumab concentrations and clinical responses. These analyses suggest that no dose adjustments are necessary in patients with CSU for age (12-75 years), race/ethnicity, gender, body weight, body mass index, baseline IgE, anti-FcRI autoantibodies or concomitant use of H2 antihistamines or LTRAs.
Renal and hepatic impairment
There are no pharmacokinetic or pharmacodynamic data in allergic asthma or CSU patients with renal or hepatic impairment (see sections 4.2 and 4.4).
Preclinical safety data
The safety of omalizumab has been studied in the cynomolgus monkey, since omalizumab binds to cynomolgus and human IgE with similar affinity. Antibodies to omalizumab were detected in some monkeys following repeated subcutaneous or intravenous administration. However, no apparent toxicity, such as immune complex-mediated disease or complement-dependent cytotoxicity, was seen. There was no evidence of an anaphylactic response due to mast-cell degranulation in cynomolgus monkeys.
Chronic administration of omalizumab at dose levels of up to 250 mg/kg (at least 14 times the highest recommended clinical dose in mg/kg according to the recommended dosing table) was well tolerated in non-human primates (both adult and juvenile animals), with the exception of a dose-related and age-dependent decrease in blood platelets, with a greater sensitivity in juvenile animals. The serum concentration required to attain a 50% drop in platelets from baseline in adult cynomolgus monkeys was roughly 4- to 20-fold higher than anticipated maximum clinical serum concentrations. In addition, acute haemorrhage and inflammation were observed at injection sites in cynomolgus monkeys.
Formal carcinogenicity studies have not been conducted with omalizumab.
In reproduction studies in cynomolgus monkeys, subcutaneous doses up to 75 mg/kg per week (at least 8 times the highest recommended clinical dose in mg/kg over a 4-week period) did not elicit maternal toxicity, embryotoxicity or teratogenicity when administered throughout organogenesis and did not elicit adverse effects on foetal or neonatal growth when administered throughout late gestation, delivery and nursing.
Omalizumab is excreted in breast milk in cynomolgus monkeys. Milk levels of omalizumab were 0.15% of the maternal serum concentration.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.