XOSPATA Film-coated tablet Ref.[109315] Active ingredients: Gilteritinib
Source: European Medicines Agency (EU) Revision Year: 2023 Publisher: Astellas Pharma Europe B.V., Sylviusweg 62, 2333 BE Leiden, The Netherlands
5.1. Pharmacodynamic properties
Pharmacotherapeutic group: antineoplastic agents, protein kinase inhibitors
ATC code: L01EX13
Mechanism of action
Gilteritinib fumarate is a FLT3 and AXL inhibitor.
Gilteritinib inhibits FLT3 receptor signalling and proliferation in cells exogenously expressing FLT3 including FLT3-ITD, FLT3-D835Y, and FLT3-ITD-D835Y, and it induces apoptosis in leukemic cells expressing FLT3-ITD.
Pharmacodynamic effects
In patients with relapsed or refractory AML receiving gilteritinib 120 mg, substantial (>90%) inhibition of FLT3 phosphorylation was rapid (within 24 hours after first dose) and sustained, as characterised by an ex vivo plasma inhibitory activity (PIA) assay.
Prolonged QT interval
A concentration-related increase in change from baseline of QTcF was observed across gilteritinib doses ranging from 20 to 450 mg. The predicted mean change from baseline of QTcF at the mean steady-state Cmax (282.0 ng/mL) at the 120 mg daily dose was 4.96 msec with an upper 1-sided 95% CI = 6.20 msec.
Clinical efficacy and safety
Relapsed or refractory AML
Efficacy and safety were evaluated in the active-controlled, phase 3 study (2215-CL-0301). ADMIRAL study (2215-CL-0301) The ADMIRAL study is a Phase 3, open-label, multicentre, randomised clinical study of adult patients with relapsed or refractory AML with a FLT3 mutation as determined by the LeukoStrat CDx FLT3 Mutation Assay. In this study, 371 patients were randomised in a 2:1 ratio to receive gilteritinib or one of the following salvage chemotherapies (247 in the gilteritinib arm and 124 in the salvage chemotherapy arm):
- cytarabine 20 mg twice daily by subcutaneous injection (SC) or intravenous infusion (IV) for 10 days (days 1 through 10) (LoDAC)
- azacitidine 75 mg/m² once daily by SC or IV for 7 days (days 1 through 7)
- mitoxantrone 8 mg/m², etoposide 100 mg/m² and cytarabine 1000 mg/m² once daily by IV for 5 days (days 1 through 5) (MEC)
- granulocyte colony-stimulating factor 300 mcg/m² once daily by SC for 5 days (days 1 to 5), fludarabine 30 mg/m² once daily by IV for 5 days (days 2 through 6), cytarabine 2000 mg/m² once daily by IV for 5 days (days 2 through 6), idarubicin 10 mg/m² once daily by IV for 3 days (days 2 through 4) (FLAG-Ida).
Patients included were relapsed or refractory after first line AML therapy and were stratified by response to prior AML treatment and preselected chemotherapy i.e. high or low intensity. While the study included patients with various AML-related cytogenetic abnormalities, patients with acute promyelocytic leukaemia (APL) or therapy-related AML were excluded.
Sixteen patients were randomised but not treated in the study (1 patient in the gilteritinib arm and 15 patients in the chemotherapy arm). Gilteritinib was given orally at a starting dose of 120 mg daily until unacceptable toxicity or lack of clinical benefit. Dose reductions were allowed, to manage adverse reactions, and dose increases were allowed, for those patients who did not respond at the starting dose of 120 mg.
Of the patients who were pre-selected to receive salvage chemotherapy, 60.5% were randomised to high intensity and 39.5% to low intensity. MEC and FLAG-Ida were given for up to two cycles depending on response to first cycle. LoDAC and azacitidine were given in continuous 4-week cycles until unacceptable toxicity or lack of clinical benefit.
The demographic and baseline characteristics were well-balanced between the two treatment arms. The median age at randomisation was 62 years (range 20 to 84 years) in the gilteritinib arm and 62 years (range 19 to 85 years) in the salvage chemotherapy arm. In the study 42% of patients were 65 years or older and 12% were 75 years or older. Fifty-four percent of the patients were female. Most patients in the study were Caucasian (59.3%); 27.5% Asian, 5.7% Black, 4% other races and 3.5% unknown. The majority of patients (83.8%) had an ECOG performance status score of 0 or 1. Patients had the following confirmed mutations: FLT3-ITD alone (88.4%), FLT3-TKD alone (8.4%) or both FLT3-ITD and FLT3-TKD (1.9%). Twelve percent of patients received previous treatment with another FLT3 inhibitor. A majority of patients had AML with intermediate risk cytogenetics (73%), 10% had unfavourable, 1.3% had favourable and 15.6% had unclassified cytogenetics.
Prior to treatment with gilteritinib, 39.4% of patients had primary refractory AML and the majority of these patients were classified as refractory after 1 cycle of chemotherapy induction treatment, 19.7% had relapsed AML after an allogeneic haematopoietic stem cell transplant (HSCT) and 41% had relapsed AML with no allogeneic HSCT.
The primary efficacy endpoint for the final analysis was OS in the intent-to-treat (ITT) population, measured from the date of randomisation until death by any cause (number of events analysed was 261). Patients randomised to the gilteritinib arm had significantly longer survival compared to the chemotherapy arm (HR 0.637; 95% CI 0.490–0.830; 1 sided p-value: 0.0004). The median OS was 9.3 months for patients receiving gilteritinib and 5.6 months for those receiving chemotherapy. Efficacy was further supported by the rate of complete remission (CR)/complete remission with partial haematologic recovery (CRh) (Table 3, Figure 1).
Table 3. ADMIRAL study overall survival and complete remission in patients with relapsed or refractory AML:
| Gilteritinib (N=247) | Chemotherapy (N=124) | |
|---|---|---|
| Overall survival | ||
| Deaths, n (%) | 171 (69.2) | 90 (72.6) |
| Median in months (95% CI) | 9.3 (7.7, 10.7) | 5.6 (4.7, 7.3) |
| Hazard Ratio (95% CI) | 0.637 (0.490, 0.830) | |
| p-value (1-sided) | 0.0004 | |
| 1 year survival rate, % (95% CI) | 37.1 (30.7, 43.6) | 16.7 (9.9, 25) |
| Complete remission | ||
| CRa (95% CIb) | 21.1% (16.1, 26.7) | 10.5% (5.7, 17.3) |
| CRhc (95% CIb) | 13% (9, 17.8) | 4.8% (1.8, 10.2) |
| CR/CRh (95% CIb) | 34% (28.1, 40.3) | 15.3% (9.5, 22.9) |
CI: confidence interval
a CR was defined as an absolute neutrophil count ≥1.0 x 109/L, platelets ≥100 x 109/L, normal marrow differential with <5% blasts, must have been red blood cells, platelet transfusion independent and no evidence of extramedullary leukemia.
b The 95% CI rate was calculated using the exact method based on binomial distribution.
c CRh was defined as marrow blasts <5%, partial haematologic recovery absolute neutrophil count ≥0.5 x 109/L and platelets ≥50 x 109/L, no evidence of extramedullary leukemia and could not have been classified as CR.
Figure 1. Kaplan-Meier plot of overall survival in ADMIRAL study:
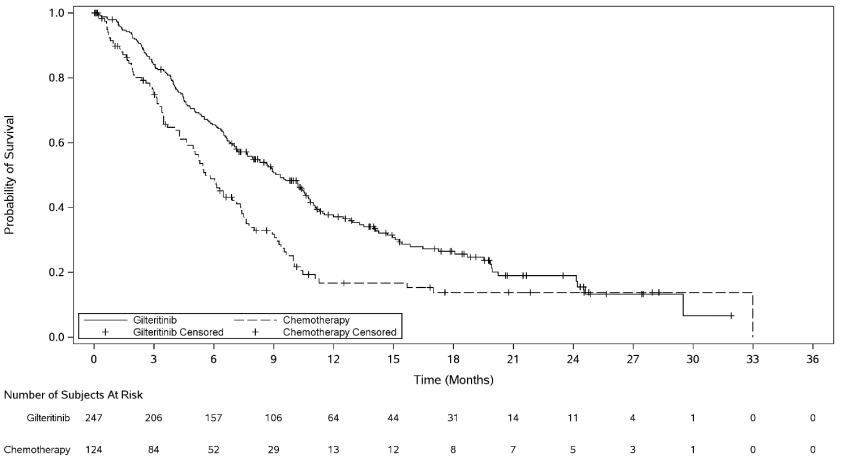
For patients who achieved a CR/CRh, the median time to first response was 3.7 months (range, 0.9 to 10.6 months) in the gilteritinib arm and 1.2 months (range: 1 to 2.6 months) in the salvage chemotherapy arm. The median time to best response of CR/CRh was 3.8 months (range, 0.9 to 16 months) in the gilteritinib arm and 1.2 months (range: 1 to 2.6 months) in the salvage chemotherapy arm.
CHRYSALIS study (2215-CL-0101)
The supportive Phase ½ dose-escalation study 2215-CL-0101 included 157 patients with FLT3 mutated AML treated with either 1 or >1 prior lines of treatment in the combined dose group (i.e. 80 mg, 120 mg or 200 mg); 31.2% received 1 prior line of treatment and 68.8% received >1 prior lines of treatment.
The response rate (CR/CRh) observed in Study 2215-CL-0101 in the patients who received more than 1 line of prior therapy was 21.4% and 15.7% for the 120 mg dose and the combined dose levels, respectively. The median OS was 7.2 months and 7.1 months for the 120 mg dose and the combined dose levels, respectively.
Paediatric population
The European Medicines Agency has deferred the obligation to submit the results of studies with Xospata in one or more subsets of the paediatric population in the treatment of acute myeloid leukaemia. See 4.2 for information on paediatric use.
5.2. Pharmacokinetic properties
Absorption
Following oral administration of gilteritinib, peak plasma concentrations are observed at a median tmax approximately between 4 and 6 hours in healthy volunteers and patients with relapsed or refractory AML. Gilteritinib undergoes first-order absorption with an estimated absorption rate (ka) of 0.43 h-1 with a lag time of 0.34 hours based on population PK modelling. Median steady-state maximum concentration (Cmax) is 282.0 ng/mL (CV% = 50.8), and area under the plasma concentration curve during 24-hour dosing interval (AUC0-24) is 6180 ng·h/mL (CV% = 46.4) after once-daily dosing of 120 mg gilteritinib. Steady-state plasma levels are reached within 15 days of once-daily dosing with an approximate 10-fold accumulation.
Effect of food
In healthy adults, gilteritinib Cmax and AUC decreased by approximately 26% and less than 10%, respectively, when a single 40 mg dose of gilteritinib was co-administered with a high fat meal compared to gilteritinib exposure in fasted state. Median tmax was delayed 2 hours when gilteritinib was administered with a high-fat meal.
Distribution
The population estimate of central and peripheral volume of distribution were 1092 L and 1100 L, respectively. These data indicate gilteritinib distributes extensively outside of plasma, which may indicate extensive tissue distribution. In vivo plasma protein binding in humans is approximately 90% and gilteritinib is primarily bound to albumin.
Biotransformation
Based on in vitro data, gilteritinib is primarily metabolised via CYP3A4. The primary metabolites in humans include M17 (formed via N-dealkylation and oxidation), M16 and M10 (both formed via N-dealkylation) and were observed in animals. None of these three metabolites exceeded 10% of overall parent exposure. The pharmacological activity of the metabolites against FLT3 and AXL receptors is unknown.
Transporter drug-drug interactions
In vitro experiments demonstrated that gilteritinib is a substrate of P-gp and BCRP. Gilteritinib may potentially inhibit BCRP, P-gp and OCT1 at clinically relevant concentrations (see section 4.5).
Elimination
After a single dose of [14C]-gilteritinib, gilteritinib is primarily excreted in faeces with 64.5% of the total administered dose recovered in faeces. Approximately 16.4% of the total dose was excreted in urine as unchanged drug and metabolites. Gilteritinib plasma concentrations declined in a bi-exponential manner with a population mean estimated half-life of 113 hours. The estimated apparent clearance (CL/F) based on the population PK model is 14.85 L/h.
Linearity/non-linearity
In general, gilteritinib exhibited linear, dose-proportional pharmacokinetics after single and multiple dose administration at doses ranging from 20 to 450 mg in patients with relapsed or refractory AML.
Special populations
A population pharmacokinetic analysis was performed to evaluate the impact of intrinsic and extrinsic covariates on the predicted exposure of gilteritinib in patients with relapsed or refractory AML. Covariate analysis indicated that age (20 years to 90 years), and body weight (36 kg to 157 kg) were statistically significant; however predicted change in gilteritinib exposure was less than 2-fold.
Hepatic impairment
The effect of hepatic impairment on gilteritinib pharmacokinetics was studied in subjects with mild (Child-Pugh Class A) and moderate (Child-Pugh Class B) hepatic impairment. Results indicate unbound gilteritinib exposure in subjects with mild or moderate hepatic impairment is comparable to that observed in subjects with normal hepatic function. The effect of mild hepatic impairment [as defined by NCI-ODWG] on gilteritinib exposure was also assessed using the population PK model and the results demonstrate little difference in predicted steady-state gilteritinib exposure relative to a typical patient with relapsed or refractory AML and normal liver function.
Gilteritinib has not been studied in patients with severe hepatic impairment (Child-Pugh Class C).
Renal impairment
The pharmacokinetics of gilteritinib were evaluated in five subjects with severe (CrCL 15 - <30 mL/min) renal impairment and in four subjects with end stage renal disease (CrCL <15 mL/min). A 1.4-fold increase in mean Cmax and 1.5-fold increase in mean AUCinf of gilteritinib was observed in subjects with severe renal impairment or end stage renal disease compared to subjects with normal renal function (n=8) (see sections 4.2 and 4.4).
5.3. Preclinical safety data
Adverse reactions not observed in clinical studies, but seen in animals (safety pharmacology/repeat dose toxicity) at exposure levels similar to clinical exposure levels and with possible relevance to clinical use were as follows.
Safety pharmacology
In rats, decreased urination at 30 mg/kg and higher and decreased defecation at 100 mg/kg were observed. In dogs, positive faecal occult blood at 10 mg/kg and higher, a decrease in the blood calcium concentration at 30 mg/kg, and salivation and an increase followed by a decrease in the blood calcium concentration at 100 mg/kg were observed. These changes were observed at plasma exposure levels similar to or less than clinical exposure levels. A possible clinical relevance of these findings is unknown.
Repeat dose toxicity
In the repeated dose toxicity studies in rats and dogs, target organs of toxicity were the gastrointestinal tract (heamorrhage in dogs), lymphohaematopoietic system (lymphocyte necrosis and bone marrow hypocellularity with changes in haematological parameters), eye (inflammation and lens opacity in rats, fundus colour change in dogs, retinal vacuolation), lung (interstitial pneumonia in rats and inflammation in dogs), kidney (renal tubule changes with a positive urine occult blood reaction) and liver (hepatocyte vacuolation), urinary bladder (epithelial vacuolation), epithelial tissue (ulcer and inflammation), and phospholipidosis (lung and kidney in rats). These changes were observed at plasma exposure levels similar to or less than clinical exposure levels. Reversibility of most of the changes was indicated by the end of the 4-week recovery period. A possible clinical relevance of these findings is unknown.
Genotoxicity
Gilteritinib did not induce gene mutation or chromosomal aberrations in vitro. The in vivo micronucleus test showed that gilteritinib has a potential to induce micronuclei in mice.
Reproductive toxicity
Gilteritinib showed suppressed foetal growth, and induced embryo-foetal deaths and teratogenicity in the embryo-foetal development studies in rats at exposure levels similar to clinical exposure levels. Placental transfer of gilteritinib was shown in the rat resulting in transfer of radioactivity to the foetus similar to that observed in maternal plasma.
Gilteritinib was excreted into the milk of lactating rats with milk concentrations being higher than in maternal plasma. Gilteritinib was distributed through the breast milk to different tissues, except for the brain, of suckling rats.
Juvenile animal toxicity study
In the juvenile toxicity study in rats, the minimum lethal dose level (2.5 mg/kg/day) was much lower than that of adult rats (20 mg/kg/day). The gastrointestinal tract was identified as one of the target organs similar as in adult rats.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.