XTANDI Soft capsule Ref.[9661] Active ingredients: Enzalutamide
Source: European Medicines Agency (EU) Revision Year: 2024 Publisher: Astellas Pharma Europe B.V., Sylviusweg 62, 2333 BE Leiden, The Netherlands
Pharmacodynamic properties
Pharmacotherapeutic group: hormone antagonists and related agents, anti-androgens
ATC code: L02BB04
Mechanism of action
Prostate cancer is known to be androgen sensitive and responds to inhibition of androgen receptor signalling. Despite low or even undetectable levels of serum androgen, androgen receptor signalling continues to promote disease progression. Stimulation of tumour cell growth via the androgen receptor requires nuclear localization and DNA binding. Enzalutamide is a potent androgen receptor signalling inhibitor that blocks several steps in the androgen receptor signalling pathway. Enzalutamide competitively inhibits androgen binding to androgen receptors, and consequently; inhibits nuclear translocation of activated receptors and inhibits the association of the activated androgen receptor with DNA even in the setting of androgen receptor overexpression and in prostate cancer cells resistant to anti-androgens. Enzalutamide treatment decreases the growth of prostate cancer cells and can induce cancer cell death and tumour regression. In preclinical studies enzalutamide lacks androgen receptor agonist activity.
Pharmacodynamic effects
In a phase 3 clinical trial (AFFIRM) of patients who failed prior chemotherapy with docetaxel, 54% of patients treated with enzalutamide, versus 1.5% of patients who received placebo, had at least a 50% decline from baseline in PSA levels.
In another phase 3 clinical trial (PREVAIL) in chemo-naïve patients, patients receiving enzalutamide demonstrated a significantly higher total PSA response rate (defined as a ≥50% reduction from baseline), compared with patients receiving placebo, 78.0% versus 3.5% (difference = 74.5%, p<0.0001).
In a phase 2 clinical trial (TERRAIN) in chemo-naïve patients, patients receiving enzalutamide demonstrated a significantly higher total PSA response rate (defined as a ≥50% reduction from baseline), compared with patients receiving bicalutamide, 82.1% versus 20.9% (difference = 61.2%, p<0.0001).
In a single arm trial (9785-CL-0410) of patients previously treated with at least 24 weeks of abiraterone (plus prednisone), 22.4% had a ≥50% decrease from baseline in PSA levels. According to prior chemotherapy history, the results proportion of patients with a ≥50% decrease in PSA levels were 22.1% and 23.2%, for the no prior chemotherapy and prior chemotherapy patient groups, respectively.
In the MDV3100-09 clinical trial (STRIVE) of non-metastatic and metastatic CRPC, patients receiving enzalutamide demonstrated a significantly higher total confirmed PSA response rate (defined as a ≥50% reduction from baseline) compared with patients receiving bicalutamide, 81.3% versus 31.3% (difference = 50.0%, p<0.0001).
In the MDV3100-14 clinical trial (PROSPER) of non-metastatic CRPC, patients receiving enzalutamide demonstrated a significantly higher confirmed PSA response rate (defined as a ≥50% reduction from baseline), compared with patients receiving placebo, 76.3% versus 2.4% (difference = 73.9%, p<0.0001).
Clinical efficacy and safety
Efficacy of enzalutamide was established in three randomised placebo-controlled multicentre phase 3 clinical studies [MDV3100-14 (PROSPER), CRPC2 (AFFIRM), MDV3100-03 (PREVAIL)] of patients with progressive prostate cancer who had disease progression on androgen deprivation therapy [LHRH analogue or after bilateral orchiectomy]. The PREVAIL study enrolled metastatic CRPC chemotherapy-naïve patients; whereas the AFFIRM study enrolled metastatic CRPC patients who had received prior docetaxel; and the PROSPER study enrolled patients with non-metastatic CRPC. Efficacy in patients with mHSPC was established in one randomised, placebo-controlled multicentre phase 3 clinical study [9785-CL-0335 (ARCHES)]. Another randomised, placebo-controlled multicentre phase 3 clinical study [MDV3100-13 (EMBARK)] established efficacy in patients with high-risk BCR nmHSPC. All patients were treated with a LHRH analogue or had bilateral orchiectomy, unless otherwise indicated.
In the active treatment arms, Xtandi was administered orally at a dose of 160 mg daily. In the five clinical studies (EMBARK, ARCHES, PROSPER, AFFIRM and PREVAIL), patients received placebo in the control arm and patients were not required to take prednisone.
Changes in PSA serum concentration independently do not always predict clinical benefit. Therefore, in the five studies it was recommended that patients be maintained on their study treatments until suspension or discontinuation criteria were met as specified below for each study.
MDV3100-13 (EMBARK) Study (patients with high-risk BCR non-metastatic HSPC)
The EMBARK study enrolled 1068 patients with high-risk BCR nmHSPC who were randomised 1:1:1 to receive treatment with enzalutamide orally at a dose of 160 mg once daily concurrently with ADT (N=355), enzalutamide orally at a dose of 160 mg once daily as open-label monotherapy (N=355), or placebo orally once daily concurrently with ADT (N=358) (ADT defined as leuprolide). All patients had prior definitive therapy with radical prostatectomy or radiotherapy (including brachytherapy) or both, with curative intent. Patients were required to have confirmation of non-metastatic disease by blinded independent central review (BICR), and high-risk biochemical recurrence (defined by a PSA doubling time ≤9 months). Patients were also required to have PSA values ≥1 ng/mL if they had prior radical prostatectomy (with or without radiotherapy) as the primary treatment for prostate cancer, or PSA values at least 2 ng/mL above the nadir if they had prior radiotherapy only. Patients who had a prior prostatectomy and were suitable candidates for salvage radiotherapy as determined by the investigator were excluded from the study.
Patients were stratified by screening PSA (≤10 ng/mL vs. >10 ng/mL), PSA doubling time (≤3 months versus >3 months to ≤9 months), and prior hormonal therapy (prior hormonal therapy vs. no prior hormonal therapy). For patients whose PSA values were undetectable (<0.2 ng/mL) at week 36, treatment was suspended at week 37 and then reinitiated when PSA values increased to ≥2.0 ng/mL for patients with prior prostatectomy or ≥5.0 ng/mL for patients without prior prostatectomy. For patients whose PSA values were detectable at week 36 (≥0.2 ng/mL), treatment continued without suspension until permanent treatment discontinuation criteria were met. Treatment was permanently discontinued when development of radiographic progression was confirmed by central review after the initial local read.
The demographic and baseline characteristics were well balanced between the three treatment groups. The overall median age at randomisation was 69 years (range: 49.0 – 93.0). Most patients in the total population were Caucasian (83.2%), 7.3% were Asian, and 4.4% were Black. The median PSA doubling time was 4.9 months. Seventy-four percent of patients had prior definitive therapy with radical prostatectomy, 75% of patients had prior therapy with radiotherapy (including brachytherapy), and 49% of patients had prior therapy with both. Thirty-two percent of patients had a Gleason score of ≥8. The Eastern Cooperative Oncology Group Performance Status (ECOG PS) score was 0 for 92% of patients and 1 for 8% of patients at study entry.
Metastasis-free survival (MFS) in patients randomised to receive enzalutamide plus ADT compared to patients randomised to receive placebo plus ADT was the primary endpoint. Metastasis-free survival was defined as the time from randomisation to radiographic progression or death on study, whichever occurred first.
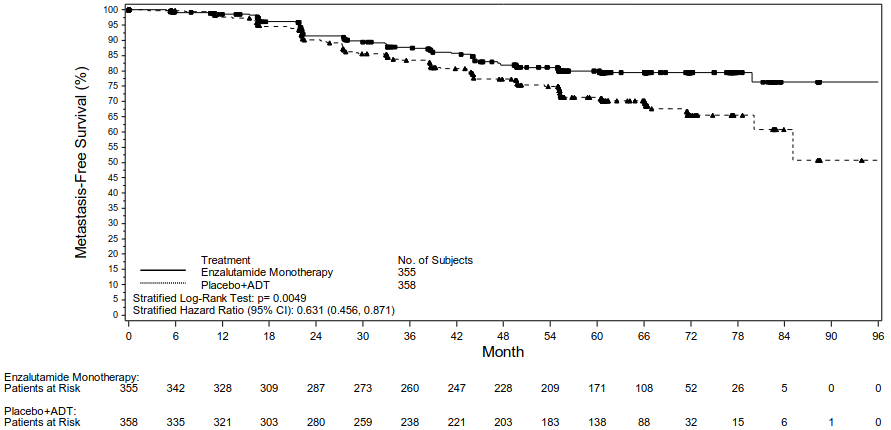
Multiplicity tested secondary endpoints that were assessed were time to PSA progression, time to first use of antineoplastic therapy, and overall survival. Another multiplicity tested secondary endpoint was MFS in patients randomised to receive enzalutamide as monotherapy compared to patients randomised to receive placebo plus ADT.
Enzalutamide plus ADT and as monotherapy demonstrated a statistically significant improvement in MFS as compared to placebo plus ADT. Key efficacy results are presented in Table 2.
Table 2. Summary of efficacy in patients treated with either enzalutamide plus ADT, placebo plus ADT, or enzalutamide as monotherapy, in the EMBARK study (intent-to-treat analysis):
| Enzalutamide plus ADT (N=355) | Placebo plus ADT (N=358) | Enzalutamide as Monotherapy (N=355) | |
|---|---|---|---|
| Metastasis-free Survival1 | |||
| Number of events (%)2 | 45 (12.7) | 92 (25.7) | 63 (17.7) |
| Median, months (95% CI)3 | NR (NR, NR) | NR (85.1, NR) | NR (NR, NR) |
| Hazard ratio relative to Placebo plus ADT (95% CI)4 | 0.42 (0.30, 0.61) | -- | 0.63 (0.46, 0.87) |
| P-value for comparison to Placebo plus ADT5 | p<0.0001 | -- | p=0.0049 |
| Time to PSA Progression6 | |||
| Number of events (%)2 | 8 (2.3) | 93 (26.0) | 37 (10.4) |
| Median, months (95% CI)3 | NR (NR, NR) | NR (NR, NR) | NR (NR, NR) |
| Hazard ratio relative to Placebo plus ADT (95% CI)4 | 0.07 (0.03, 0.14) | -- | 0.33 (0.23, 0.49) |
| P-value for comparison to Placebo plus ADT5 | p<0.0001 | -- | p<0.0001 |
| Time to Start of New Antineoplastic Therapy | |||
| Number of events (%)7 | 58 (16.3) | 140 (39.1) | 84 (23.7) |
| Median, months (95% CI)3 | NR (NR, NR) | 76.2 (71.3, NR) | NR (NR, NR) |
| Hazard ratio relative to Placebo plus ADT (95% CI)4 | 0.36 (0.26, 0.49) | -- | 0.54 (0.41, 0.71) |
| P-value for comparison to Placebo plus ADT5 | p<0.0001 | -- | p<0.0001 |
| Overall Survival8 | |||
| Number of events (%) | 33 (9.3) | 55 (15.4) | 42 (11.8) |
| Median, months (95% CI)3 | NR (NR, NR) | NR (NR, NR) | NR (NR, NR) |
| Hazard ratio relative to Placebo plus ADT (95% CI)4 | 0.59 (0.38, 0.91) | -- | 0.78 (0.52, 1.17) |
| P-value for comparison to Placebo plus ADT5 | p=0.01539 | -- | p=0.23049 |
NR = Not reached.
1 Median follow-up time of 61 months.
2 Based on the earliest contributing event (radiographic progression or death).
3 Based on Kaplan-Meier estimates.
4 Hazard Ratio is based on a Cox regression model stratified by screening PSA, PSA doubling time, and prior hormonal therapy.
5 Two-sided P-value is based on a stratified log-rank test by screening PSA, PSA doubling time, and prior hormonal therapy.
6 Based on the PSA Progression compliant with Prostate Cancer Clinical Trials Working Group 2 criteria.
7 Based on the first postbaseline use of antineoplastic therapy for prostate cancer.
8 Based upon a pre-specified interim analysis with data cutoff date of 31 Jan 2023 and a median follow-up time of
65 months.
9 The result did not meet the pre-specified two-sided significance level of p≤0.0001.
Figure 1. Kaplan-Meier curves of MFS in the Enzalutamide plus ADT vs. Placebo plus ADT treatment arms of the EMBARK study (intent-to-treat analysis):
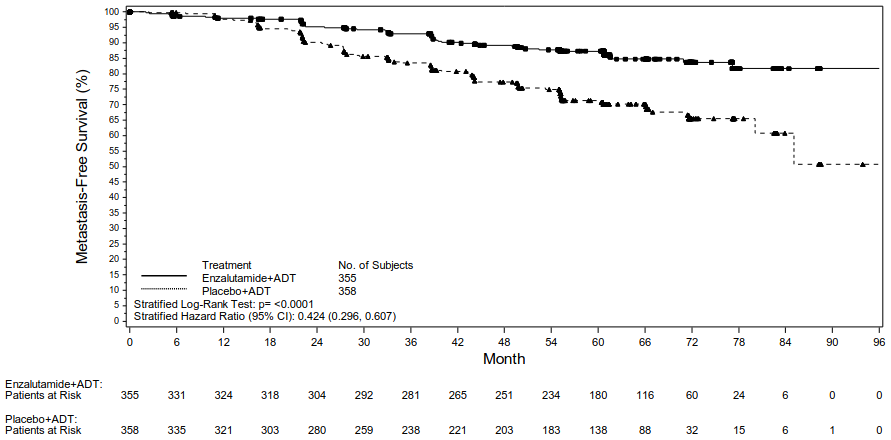
Figure 2. Kaplan-Meier curves of MFS in the Enzalutamide as Monotherapy vs. Placebo plus ADT treatment arms of the EMBARK study (intent-to-treat analysis):
Following the administration of ADT as enzalutamide plus ADT or placebo plus ADT, testosterone levels rapidly decreased to castrate levels and remained low until treatment interruption at 37 weeks. Following the interruption, testosterone levels gradually rose to near-baseline levels. Upon re-initiation of treatment, they fell again to castrate levels. In the enzalutamide as monotherapy arm, testosterone levels increased after treatment initiation and returned towards baseline levels upon treatment interruption. They increased once again after treatment with enzalutamide was re-initiated.
9785-CL-0335 (ARCHES) Study (patients with metastatic HSPC)
The ARCHES study enrolled 1150 patients with mHSPC randomised 1:1 to receive treatment with enzalutamide plus ADT or placebo plus ADT (ADT defined as LHRH analogue or bilateral orchiectomy). Patients received enzalutamide at 160 mg once daily (N=574) or placebo (N=576).
Patients with metastatic prostate cancer documented by positive bone scan (for bone disease) or metastatic lesions on CT or MRI scan (for soft tissue) were eligible. Patients whose disease spread was limited to regional pelvic lymph nodes were not eligible. Patients were allowed to receive up to 6 cycles of docetaxel therapy with final treatment administration completed within 2 months of day 1 and no evidence of disease progression during or after the completion of docetaxel therapy. Excluded were patients with known or suspected brain metastasis or active leptomeningeal disease or with a history of seizure or any contribution that may dispose to seizure.
The demographic and baseline characteristics were well balanced between the two treatment groups. The median age at randomisation was 70 years in both treatment groups. Most patients in the total population were Caucasian (80.5%); 13.5% were Asian and 1.4% were Black. The Eastern Cooperative Oncology Group Performance Status (ECOG PS) score was 0 for 78% of patients and 1 for 22% of patients at study entry. Patients were stratified by low versus high volume of disease and prior docetaxel therapy for prostate cancer. Thirty-seven percent of patients had a low volume of disease and 63% of patients had a high volume of disease. Eighty-two percent of patients had not received prior docetaxel therapy, 2% received 1-5 cycles and 16% received 6 prior cycles. Treatment with concurrent docetaxel was not allowed.
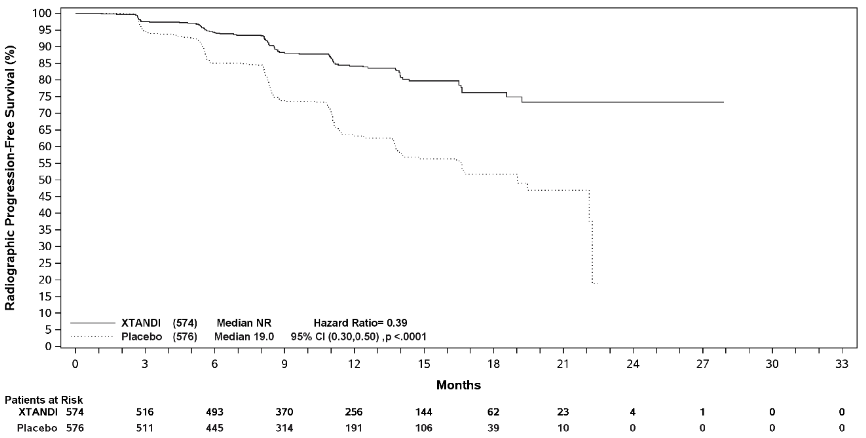
Radiographic progression-free survival (rPFS), based on independent central review, was the primary endpoint defined as the time from randomisation to the first objective evidence of radiographic disease progression or death (due to any cause from time of randomisation up until 24 weeks from study drug discontinuation), whichever occurred first.
Enzalutamide demonstrated a statistically significant 61% reduction in the risk of an rPFS event compared to placebo [HR=0.39 (95% CI: 0.30, 0.50); p<0.0001]. Consistent rPFS results were observed in patients with high or low volume of disease and patients with and without prior docetaxel therapy. The median time to an rPFS event was not reached in the enzalutamide arm and was 19.0 months (95% CI: 16.6, 22.2) in the placebo arm.
Table 3. Summary of efficacy in patients treated with either enzalutamide or placebo in the ARCHES study (intent-to-treat analysis):
| Enzalutamide plus ADT (N=574) | Placebo plus ADT (N=576) | |
|---|---|---|
| Radiographic Progression-free Survival | ||
| Number of events (%) | 91 (15.9) | 201 (34.9) |
| Median, months (95% CI)1 | NR | 19.0 (16.6, 22.2) |
| Hazard ratio (95% CI)2 | 0.39 (0.30, 0.50) | |
| P-value2 | p<0.0001 | |
NR = Not reached.
1 Calculated using Brookmeyer and Crowley method.
2 Stratified by volume of disease (low vs high) and prior docetaxel use (yes or no).
Figure 3. Kaplan-Meier curve of rPFS in ARCHES study (intent-to-treat analysis):
Key secondary efficacy endpoints assessed in the study included time to PSA progression, time to start of new antineoplastic therapy, PSA undetectable rate (decline to < 0.2 μg/L), and objective response rate (RECIST 1.1 based on independent review). Statistically significant improvements in patients treated with enzalutamide compared to placebo were demonstrated for all these secondary endpoints.
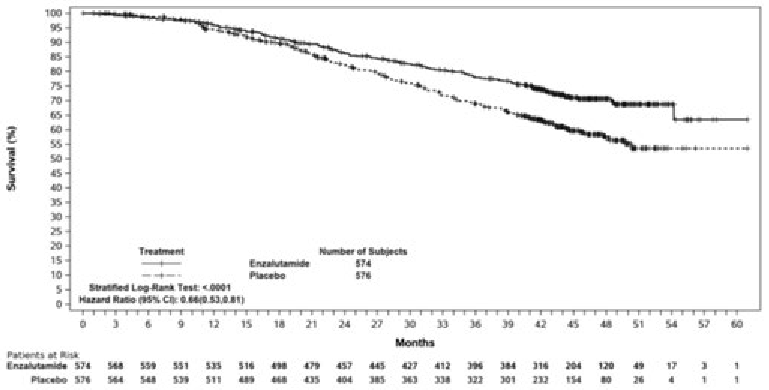
Another key secondary efficacy endpoint assessed in the study was overall survival. At the pre-specified final analysis for overall survival, conducted when 356 deaths were observed, a statistically significant 34% reduction in the risk of death was demonstrated in the group randomised to receive enzalutamide compared with the group randomised to receive placebo [HR=0.66, (95% CI: 0.53; 0.81), p<0.0001]. The median time for overall survival was not reached in either treatment group. The estimated median follow-up time for all patients was 44.6 months (see Figure 4).
Figure 4. Kaplan-Meier Curves of overall survival in the ARCHES study (intent-to-treat analysis):
MDV3100-14 (PROSPER) study (patients with non-metastatic CRPC)
The PROSPER study enrolled 1401 patients with asymptomatic, high-risk non-metastatic CRPC who continued on androgen deprivation therapy (ADT; defined as LHRH analogue or prior bilateral orchiectomy). Patients were required to have a PSA doubling time ≤ 10 months, PSA ≥ 2 ng/mL, and confirmation of non-metastatic disease by blinded independent central review (BICR).
Patients with a history of mild to moderate heart failure (NYHA Class I or II), and patients taking medicinal products associated with lowering the seizure threshold were allowed. Patients were excluded with a previous history of seizure, a condition that might predispose them to seizure, or certain prior treatments for prostate cancer (i.e., chemotherapy, ketoconazole, abiraterone acetate, aminoglutethimide and/or enzalutamide).
Patients were randomised 2:1 to receive either enzalutamide at a dose of 160 mg once daily (N = 933) or placebo (N = 468). Patients were stratified by Prostate Specific Antigen (PSA) Doubling Time (PSADT) (< 6 months or ≥ 6 months) and the use of bone-targeting agents (yes or no).
The demographic and baseline characteristics were well-balanced between the two treatment arms. The median age at randomisation was 74 years in the enzalutamide arm and 73 years in the placebo arm. Most patients (approximately 71%) in the study were Caucasian; 16% were Asian and 2% were Black. Eighty-one percent (81%) of patients had an ECOG performance status score of 0 and 19% patients had an ECOG performance status of 1.
Metastasis-free survival (MFS) was the primary endpoint defined as the time from randomisation to radiographic progression or death within 112 days of treatment discontinuation without evidence of radiographic progression, whichever occurred first. Key secondary endpoints assessed in the study were time to PSA progression, time to first use of new antineoplastic therapy (TTA), overall survival (OS). Additional secondary endpoints included time to first use of cytotoxic chemotherapy and chemotherapy-free survival. See results below (Table 4).
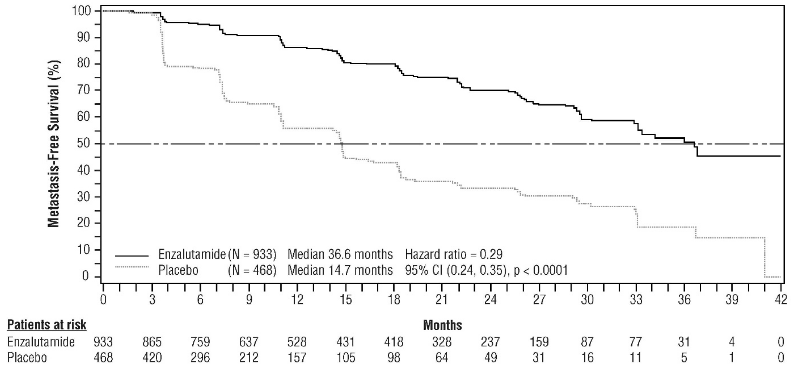
Enzalutamide demonstrated a statistically significant 71% reduction in the relative risk of radiographic progression or death compared to placebo [HR=0.29 (95% CI: 0.24, 0.35), p<0.0001]. Median MFS was 36.6 months (95% CI: 33.1, NR) on the enzalutamide arm versus 14.7 months (95% CI: 14.2, 15.0) on the placebo arm. Consistent MFS results were also observed in all pre-specified patient sub-groups including PSADT (<6 months or ≥6 months), demographic region (North America, Europe, rest of world), age (<75 or ≥75), use of a prior bone-targeting agent (yes or no) (see Figure 5).
Table 4. Summary of efficacy results in the PROSPER study (intent-to-treat analysis):
| Enzalutamide (N=933) | Placebo (N=468) | |
|---|---|---|
| Primary Endpoint | ||
| Metastasis-free survival | ||
| Number of Events (%) | 219 (23.5) | 228 (48.7) |
| Median, months (95% CI)1 | 36.6 (33.1, NR) | 14.7 (14.2, 15.0) |
| Hazard Ratio (95% CI)2 | 0.29 (0.24, 0.35) | |
| P-value3 | p<0.0001 | |
| Key Secondary Efficacy Endpoints | ||
| Overall Survival4 | ||
| Number of Events (%) | 288 (30.9) | 178 (38.0) |
| Median, months (95% CI)1 | 67.0 (64.0, NR) | 56.3 (54.4, 63.0) |
| Hazard Ratio (95% CI)2 | 0.734 (0.608, 0.885) | |
| P-value3 | p=0.0011 | |
| Time to PSA progression | ||
| Number of Events (%) | 208 (22.3) | 324 (69.2) |
| Median, months (95% CI)1 | 37.2 (33.1, NR) | 3.9 (3.8, 4.0) |
| Hazard Ratio (95% CI)2 | 0.07 (0.05, 0.08) | |
| P-value3 | p<0.0001 | |
| Time to first use of new antineoplastic therapy | ||
| Number of Events (%) | 142 (15.2) | 226 (48.3) |
| Median, months (95% CI)1 | 39.6 (37.7, NR) | 17.7 (16.2, 19.7) |
| Hazard Ratio (95% CI)2 | 0.21 (0.17, 0.26) | |
| P-value3 | p<0.0001 | |
NR = Not reached.
1 Based on Kaplan-Meier estimates.
2 HR is based on a Cox regression model (with treatment as the only covariate) stratified by PSA doubling time and prior or concurrent use of a bone targeting agent. The HR is relative to placebo with <1 favouring enzalutamide.
3. P-value is based on a stratified log-rank test by PSA doubling time (<6 months, ≥6 months) and prior or concurrent use of a bone targeting agent (yes, no).
4 Based upon a prespecified interim analysis with data cutoff date of 15 Oct 2019.
Figure 5. Kaplan-Meier Curves of metastasis-free survival in the PROSPER study (intent-to-treat analysis):
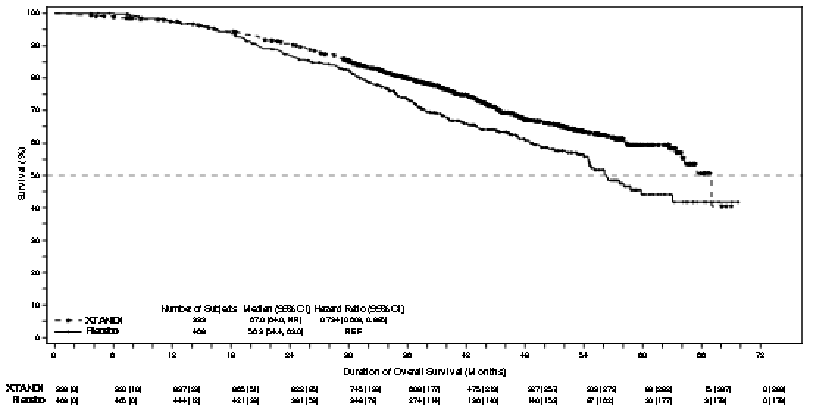
At the final analysis for overall survival, conducted when 466 deaths were observed, a statistically significant improvement in overall survival was demonstrated in patients randomised to receive enzalutamide compared with patients randomised to receive placebo with a 26.6% reduction in risk of death [hazard ratio (HR) = 0.734, (95% CI: 0.608; 0.885), p=0.0011] (see Figure 6). The median follow-up time was 48.6 and 47.2 months for the enzalutamide and placebo groups, respectively. Thirty-three percent of enzalutamide-treated and 65% of placebo-treated patients received at least one subsequent antineoplastic therapy that may prolong overall survival.
Figure 6. Kaplan-Meier Curves of overall survival in the PROSPER study (intent-to-treat analysis):
Enzalutamide demonstrated a statistically significant 93% reduction in the relative risk of PSA progression compared to placebo [HR=0.07 (95% CI: 0.05, 0.08), p<0.0001]. Median time to PSA progression was 37.2 months (95% CI: 33.1, NR) on the enzalutamide arm versus 3.9 months (95% CI: 3.8, 4.0) on the placebo arm.
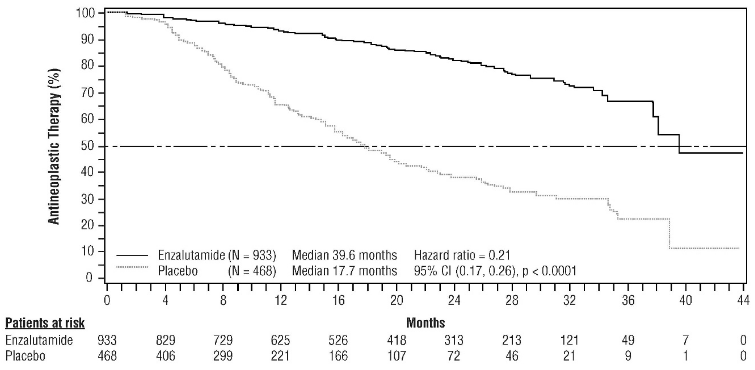
Enzalutamide demonstrated a statistically significant delay in the time to first use of new antineoplastic therapy compared to placebo [HR=0.21 (95% CI: 0.17, 0.26), p<0.0001]. Median time to first use of new antineoplastic therapy was 39.6 months (95% CI: 37.7, NR) on the enzalutamide arm versus 17.7 months (95% CI: 16.2, 19.7) on the placebo arm (see Figure 7).
Figure 7. Kaplan-Meier curves of time to first use of new antineoplastic therapy in the PROSPER study (intent-to-treat analysis):
MDV3100-09 (STRIVE) study (chemotherapy-naïve patients with non-metastatic/metastatic CRPC)
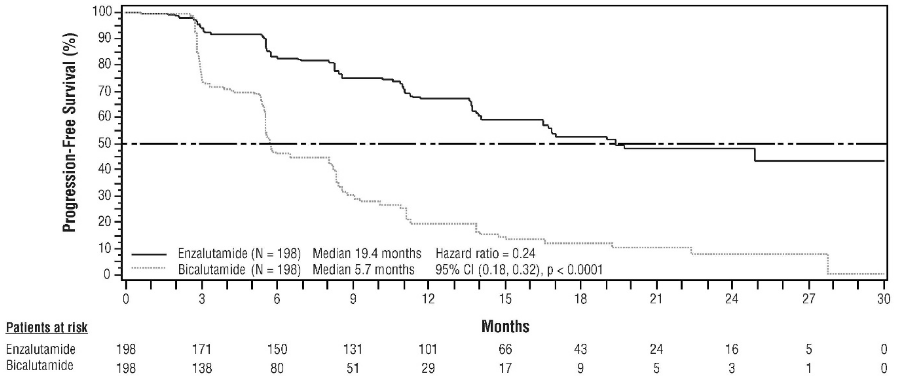
The STRIVE study enrolled 396 non-metastatic or metastatic CRPC patients who had serologic or radiographic disease progression despite primary androgen deprivation therapy who were randomised to receive either enzalutamide at a dose of 160 mg once daily (N=198) or bicalutamide at a dose of 50 mg once daily (N=198). PFS was the primary endpoint defined as the time from randomisation to the earliest objective evidence of radiographic progression, PSA progression, or death on study. Median PFS was 19.4 months (95% CI: 16.5, not reached) in the enzalutamide group versus 5.7 months (95% CI: 5.6, 8.1) in the bicalutamide group [HR=0.24 (95% CI: 0.18, 0.32), p<0.0001]. Consistent benefit of enzalutamide over bicalutamide on PFS was observed in all pre-specified patient subgroups. For the non-metastatic subgroup (N=139) a total of 19 out of 70 (27.1%) patients treated with enzalutamide and 49 out of 69 (71.0%) patients treated with bicalutamide had PFS events (68 total events). The hazard ratio was 0.24 (95% CI: 0.14, 0.42) and the median time to a PFS event was not reached in the enzalutamide group versus 8.6 months in the bicalutamide group (see Figure 8).
Figure 8. Kaplan-Meier Curves of progression-free survival in the STRIVE study (intent-to-treat analysis):
9785-CL-0222 (TERRAIN) study (chemotherapy-naïve patients with metastatic CRPC)
The TERRAIN study enrolled 375 chemo- and antiandrogen-therapy naïve patients with metastatic CRPC who were randomised to receive either enzalutamide at a dose of 160 mg once daily (N=184) or bicalutamide at a dose of 50 mg once daily (N=191). Median PFS was 15.7 months for patients on enzalutamide versus 5.8 months for patients on bicalutamide [HR=0.44 (95% CI: 0.34, 0.57), p<0.0001]. Progression-free survival was defined as objective evidence of radiographic disease progression by independent central review, skeletal-related events, initiation of new antineoplastic therapy or death by any cause, whichever occurred first. Consistent PFS benefit was observed across all pre-specified patient subgroups.
MDV3100-03 (PREVAIL) study (chemotherapy-naïve patients with metastatic CRPC)
A total of 1717 asymptomatic or mildly symptomatic chemotherapy-naïve patients were randomised 1:1 to receive either enzalutamide orally at a dose of 160 mg once daily (N = 872) or placebo orally once daily (N = 845). Patients with visceral disease, patients with a history of mild to moderate heart failure (NYHA Class I or II), and patients taking medicinal products associated with lowering the seizure threshold were allowed. Patients with a previous history of seizure or a condition that might predispose to seizure and patients with moderate or severe pain from prostate cancer were excluded. Study treatment continued until disease progression (evidence of radiographic progression, a skeletal- related event, or clinical progression) and the initiation of either a cytotoxic chemotherapy or an investigational agent, or until unacceptable toxicity.
Patient demographics and baseline disease characteristics were balanced between the treatment arms. The median age was 71 years (range 42 - 93) and the racial distribution was 77% Caucasian, 10% Asian, 2% Black and 11% other or unknown races. Sixty-eight percent (68%) of patients had an ECOG performance status score of 0 and 32% patients had an ECOG performance status of 1. Baseline pain assessment was 0 - 1 (asymptomatic) in 67% of patients and 2 - 3 (mildly symptomatic) in 32% of patients as defined by the Brief Pain Inventory Short Form (worst pain over past 24 hours on a scale of 0 to 10). Approximately 45% of patients had measurable soft tissue disease at study entry, and 12% of patients had visceral (lung and/or liver) metastases.
Co-primary efficacy endpoints were overall survival and radiographic progression-free survival (rPFS). In addition to the co-primary endpoints, benefit was also assessed using time to initiation of cytotoxic chemotherapy, best overall soft tissue response, time to first skeletal-related event, PSA response (≥ 50% decrease from baseline), time to PSA progression, and time to FACT-P total score degradation.
Radiographic progression was assessed with the use of sequential imaging studies as defined by Prostate Cancer Clinical Trials Working Group 2 (PCWG2) criteria (for bone lesions) and/or Response Evaluation Criteria in Solid Tumors (RECIST v 1.1) criteria (for soft tissue lesions). Analysis of rPFS utilised centrally-reviewed radiographic assessment of progression.
At the pre-specified interim analysis for overall survival when 540 deaths were observed, treatment with enzalutamide demonstrated a statistically significant improvement in overall survival compared to treatment with placebo with a 29.4% reduction in risk of death [HR=0.706 (95% CI: 0.60; 0.84), p<0.0001]. An updated survival analysis was conducted when 784 deaths were observed. Results from this analysis were consistent with those from the interim analysis (Table 5). At the updated analysis 52% of enzalutamide-treated and 81% of placebo-treated patients had received subsequent therapies for metastatic CRPC that may prolong overall survival.
A final analysis of 5-year PREVAIL data showed a statistically significant increase in overall survival was maintained in patients treated with enzalutamide compared to placebo [HR=0.835, (95% CI: 0.75, 0.93); p-value=0.0008] despite 28% of patients on placebo crossing over to enzalutamide. The 5-year OS rate was 26% for the enzalutamide arm compared to 21% for the placebo arm.
Table 5. Overall survival of patients treated with either enzalutamide or placebo in the PREVAIL study (intent-to-treat analysis):
| Enzalutamide (N=872) | Placebo (N=845) | |
|---|---|---|
| Pre-specified interim analysis | ||
| Number of deaths (%) | 241 (27.6%) | 299 (35.4%) |
| Median survival, months (95% CI) | 32.4 (30.1, NR) | 30.2 (28.0, NR) |
| P-value1 | p<0.0001 | |
| Hazard ratio (95% CI)2 | 0.71 (0.60, 0.84) | |
| Updated survival analysis | ||
| Number of deaths (%) | 368 (42.2%) | 416 (49.2%) |
| Median survival, months (95% CI) | 35.3 (32.2, NR) | 31.3 (28.8, 34.2) |
| P-value1 | p=0.0002 | |
| Hazard ratio (95% CI)2 | 0.77 (0.67, 0.88) | |
| 5-year survival analysis | ||
| Number of deaths (%) | 689 (79) | 693 (82) |
| Median survival, months (95% CI) | 35.5 (33.5, 38.0) | 31.4 (28.9, 33.8) |
| P-value1 | p=0.0008 | |
| Hazard ratio (95% CI)2 | 0.835 (0.75, 0.93) | |
NR = Not reached.
1 P-value is derived from an unstratified log-rank test.
2 Hazard Ratio is derived from an unstratified proportional hazards model. Hazard ratio <1 favours enzalutamide.
Figure 9. Kaplan-Meier curves of overall survival based on 5-year survival analysis in the PREVAIL study (intent-to-treat analysis):
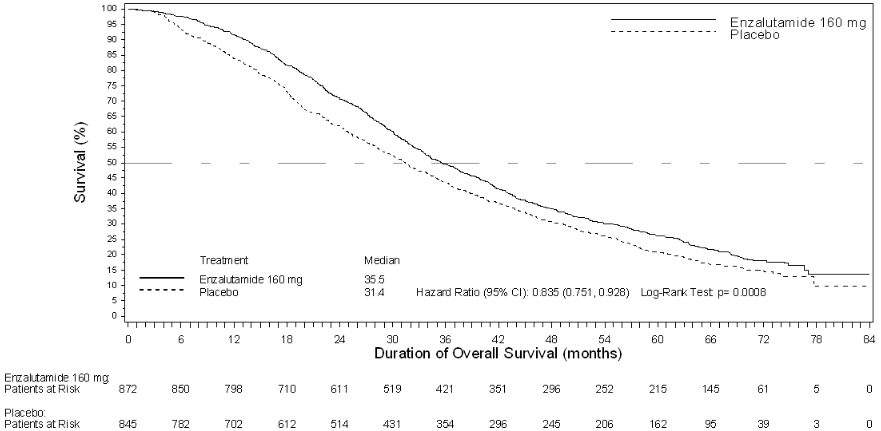
Figure 10. 5-year overall survival analysis by subgroup: Hazard ratio and 95% confidence interval in the PREVAIL study (intent-to-treat analysis):
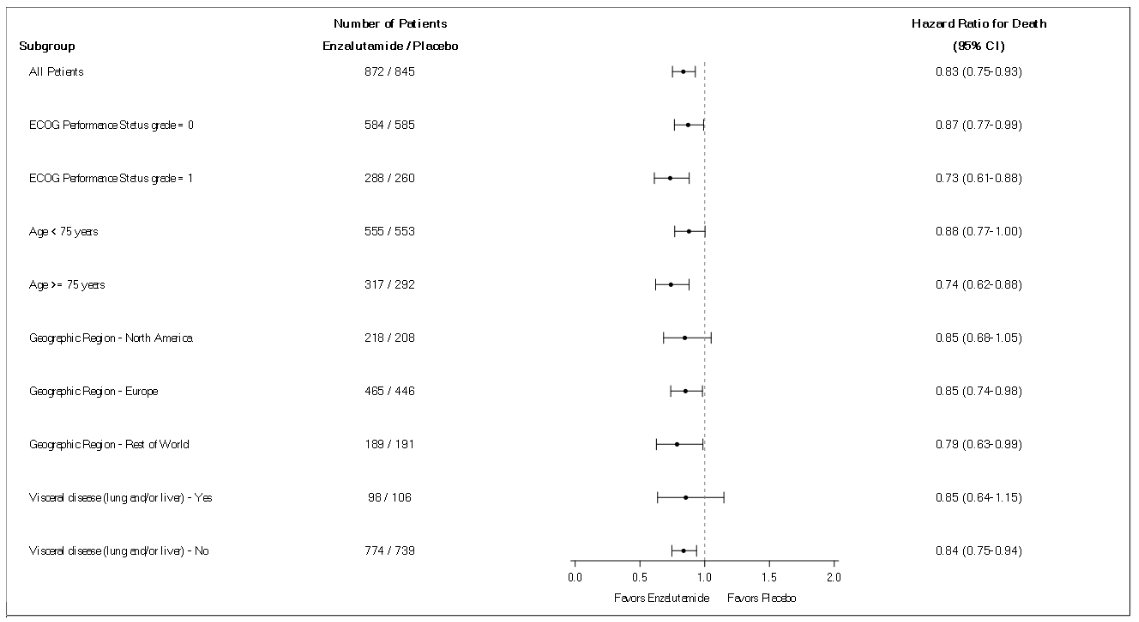
At the pre-specified rPFS analysis, a statistically significant improvement was demonstrated between the treatment groups with an 81.4% reduction in risk of radiographic progression or death [HR=0.19 (95% CI: 0.15, 0.23), p<0.0001]. One hundred and eighteen (14%) enzalutamide-treated patients and 321 (40%) of placebo-treated patients had an event. The median rPFS was not reached (95% CI: 13.8, not reached) in the enzalutamide-treated group and was 3.9 months (95% CI: 3.7, 5.4) in the placebo-treated group (Figure 11). Consistent rPFS benefit was observed across all pre-specified patient subgroups (e.g. age, baseline ECOG performance, baseline PSA and LDH, Gleason score at diagnosis, and visceral disease at screening). A pre-specified follow-up rPFS analysis based on the investigator assessment of radiographic progression demonstrated a statistically significant improvement between the treatment groups with a 69.3% reduction in risk of radiographic progression or death [HR=0.31 (95% CI: 0.27, 0.35), p<0.0001]. The median rPFS was 19.7 months in the enzalutamide group and 5.4 months in the placebo group.
Figure 11. Kaplan-Meier curves of radiographic progression-free survival in the PREVAIL study (intent-to-treat analysis):
In addition to the co-primary efficacy endpoints, statistically significant improvements were also demonstrated in the following prospectively defined endpoints.
The median time to initiation of cytotoxic chemotherapy was 28.0 months for patients receiving enzalutamide and 10.8 months for patients receiving placebo [HR=0.35 (95% CI: 0.30, 0.40), p<0.0001].
The proportion of enzalutamide-treated patients with measurable disease at baseline who had an objective soft tissue response was 58.8% (95% CI: 53.8, 63.7) compared with 5.0% (95% CI: 3.0, 7.7) of patients receiving placebo. The absolute difference in objective soft tissue response between enzalutamide and placebo arms was [53.9% (95% CI: 48.5, 59.1), p<0.0001]. Complete responses were reported in 19.7% of enzalutamide-treated patients compared with 1.0% of placebo-treated patients, and partial responses were reported in 39.1% of enzalutamide-treated patients versus 3.9% of placebo-treated patients.
Enzalutamide significantly decreased the risk of the first skeletal-related event by 28% [HR=0.718 (95% CI: 0.61, 0.84), p<0.0001]. A skeletal-related event was defined as radiation therapy or surgery to bone for prostate cancer, pathologic bone fracture, spinal cord compression, or change of antineoplastic therapy to treat bone pain. The analysis included 587 skeletal-related events, of which 389 events (66.3%) were radiation to bone, 79 events (13.5%) were spinal cord compression, 70 events (11.9%) were pathologic bone fracture, 45 events (7.6%) were change in antineoplastic therapy to treat bone pain, and 22 events (3.7%) were surgery to bone.
Patients receiving enzalutamide demonstrated a significantly higher total PSA response rate (defined as a ≥ 50% reduction from baseline), compared with patients receiving placebo, 78.0% versus 3.5% (difference = 74.5%, p<0.0001).
The median time to PSA progression per PCWG2 criteria was 11.2 months for patients treated with enzalutamide and 2.8 months for patients who received placebo [HR=0.17 (95% CI: 0.15, 0.20), p<0.0001].
Treatment with enzalutamide decreased the risk of FACT-P degradation by 37.5% compared with placebo (p<0.0001). The median time to degradation in FACT-P was 11.3 months in the enzalutamide group and 5.6 months in the placebo group.
CRPC2 (AFFIRM) study (patients with metastatic CRPC who previously received chemotherapy)
The efficacy and safety of enzalutamide in patients with metastatic CRPC who had received docetaxel and were using a LHRH analogue or had undergone orchiectomy were assessed in a randomised, placebo-controlled, multicentre phase 3 clinical trial. A total of 1199 patients were randomised 2:1 to receive either enzalutamide orally at a dose of 160 mg once daily (N=800) or placebo once daily (N=399). Patients were allowed but not required to take prednisone (maximum daily dose allowed was 10 mg prednisone or equivalent). Patients randomised to either arm were to continue treatment until disease progression (defined as confirmed radiographic progression or the occurrence of a skeletal-related event) and initiation of new systemic antineoplastic treatment, unacceptable toxicity, or withdrawal.
The following patient demographics and baseline disease characteristics were balanced between the treatment arms. The median age was 69 years (range 41 - 92) and the racial distribution was 93% Caucasian, 4% Black, 1% Asian, and 2% Other. The ECOG performance score was 0 - 1 in 91.5% of patients and 2 in 8.5% of patients; 28% had a mean Brief Pain Inventory score of ≥ 4 (mean of patient's reported worst pain over the previous 24 hours calculated for seven days prior to randomisation). Most (91%) patients had metastases in bone and 23% had visceral lung and/or liver involvement. At study entry, 41% of randomised patients had PSA progression only, whereas 59% of patients had radiographic progression. Fifty-one percent (51%) of patients were on bisphosphonates at baseline.
The AFFIRM study excluded patients with medical conditions that may predispose them to seizures (see section 4.8) and medicinal products known to decrease the seizure threshold, as well as clinically significant cardiovascular disease such as uncontrolled hypertension, recent history of myocardial infarction or unstable angina, New York Heart Association class III or IV heart failure (unless ejection fraction was ≥ 45%), clinically significant ventricular arrhythmias or AV block (without permanent pacemaker).
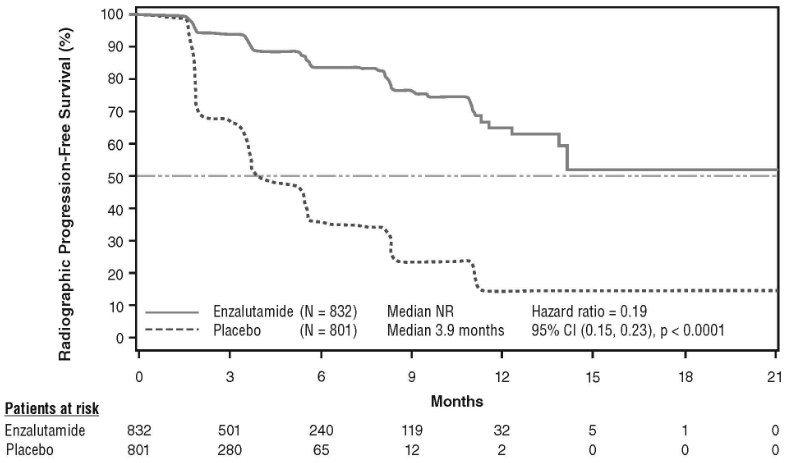
The protocol pre-specified interim analysis after 520 deaths showed a statistically significant superiority in overall survival in patients treated with enzalutamide compared to placebo (Table 6 and Figures 12 and 13).
Table 6. Overall survival of patients treated with either enzalutamide or placebo in the AFFIRM study (intent-to-treat analysis):
| Enzalutamide (N=800) | Placebo (N=399) | |
|---|---|---|
| Deaths (%) | 308 (38.5%) | 212 (53.1%) |
| Median survival (months) (95% CI) | 18.4 (17.3, NR) | 13.6 (11.3, 15.8) |
| P-value1 | p<0.0001 | |
| Hazard ratio (95% CI)2 | 0.63 (0.53, 0.75) | |
NR = Not Reached.
1 P-value is derived from a log rank test stratified by ECOG performance status score (0-1 vs. 2) and mean pain score (<4 vs. ≥4).
2 Hazard Ratio is derived from a stratified proportional hazards model. Hazard ratio <1 favours enzalutamide.
Figure 12. Kaplan-Meier curves of overall survival in the AFFIRM study (intent-to-treat analysis):
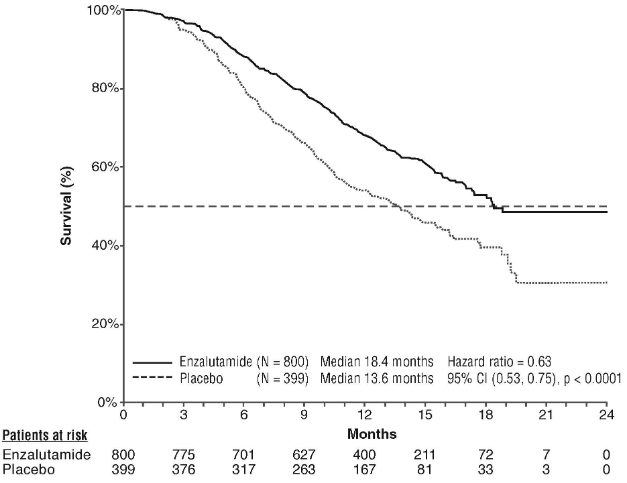
Figure 13. Overall survival by subgroup in the AFFIRM study – Hazard ratio and 95% confidence interval:
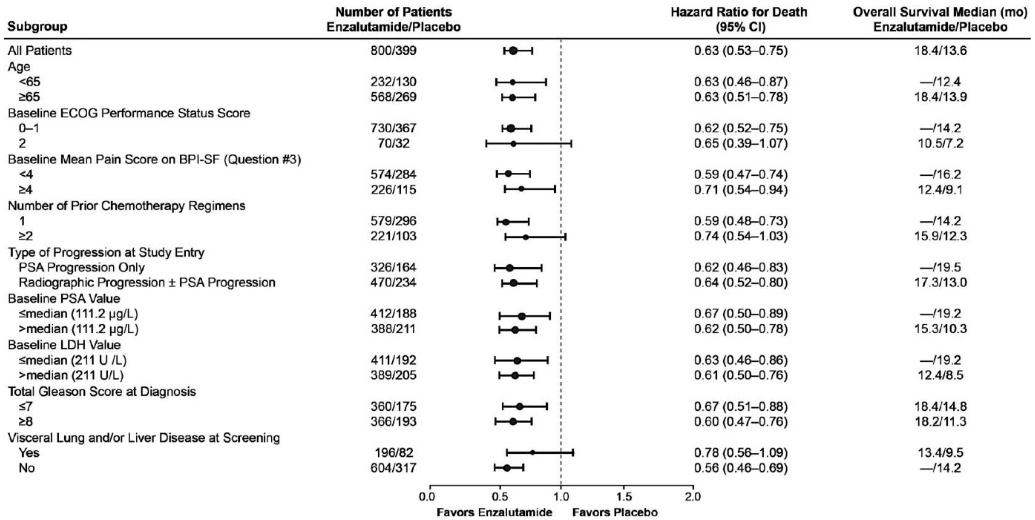
ECOG: Eastern Cooperative Oncology Group; BPI-SF: Brief Pain Inventory-Short Form; PSA: Prostate Specific Antigen
In addition to the observed improvement in overall survival, key secondary endpoints (PSA progression, radiographic progression-free survival, and time to first skeletal-related event) favoured enzalutamide and were statistically significant after adjusting for multiple testing.
Radiographic progression-free survival as assessed by the investigator using RECIST v 1.1 for soft tissue and appearance of 2 or more bone lesions in bone scan was 8.3 months for patients treated with enzalutamide and 2.9 months for patients who received placebo [HR=0.40 (95% CI: 0.35, 0.47), p<0.0001]. The analysis involved 216 deaths without documented progression and 645 documented progression events, of which 303 (47%) were due to soft tissue progression, 268 (42%) were due to bone lesion progression and 74 (11%) were due to both soft tissue and bone lesions.
Confirmed PSA decline of 50% or 90% were 54.0% and 24.8%, respectively, for patients treated with enzalutamide and 1.5% and 0.9%, respectively, for patients who received placebo (p<0.0001). The median time to PSA progression was 8.3 months for patients treated with enzalutamide and 3.0 months for patients who received placebo [HR=0.25 (95% CI: 0.20, 0.30), p<0.0001].
The median time to first skeletal-related event was 16.7 months for patients treated with enzalutamide and 13.3 months for patients who received placebo [HR=0.69 (95% CI: 0.57, 0.84), p<0.0001]. A skeletal-related event was defined as radiation therapy or surgery to bone, pathologic bone fracture, spinal cord compression or change of antineoplastic therapy to treat bone pain. The analysis involved 448 skeletal-related events, of which 277 events (62%) were radiation to bone, 95 events (21%) were spinal cord compression, 47 events (10%) were pathologic bone fracture, 36 events (8%) were change in antineoplastic therapy to treat bone pain, and 7 events (2%) were surgery to bone.
9785-CL-0410 study (enzalutamide post abiraterone in patients with metastatic CRPC)
The study was a single-arm study in 214 patients with progressing metastatic CRPC who received enzalutamide (160 mg once daily) after at least 24 weeks of treatment with abiraterone acetate plus prednisone. Median rPFS (radiologic progression free survival, the study´s primary endpoint) was 8.1 months (95% CI: 6.1, 8.3). Median OS was not reached. PSA Response (defined as ≥50% decrease from baseline) was 22.4% (95% CI: 17.0, 28.6). For the 69 patients who previously received chemotherapy, median rPFS was 7.9 months (95% CI: 5.5, 10.8). PSA Response was 23.2% (95% CI: 13.9, 34.9). For the 145 patients who had no previous chemotherapy, median rPFS was 8.1 months (95% CI: 5.7, 8.3). PSA Response was 22.1% (95% CI: 15.6, 29.7).
Although there was a limited response in some patients from treatment with enzalutamide after abiraterone, the reason for this finding is currently unknown. The study design could neither identify the patients who are likely to benefit, nor the order in which enzalutamide and abiraterone should be optimally sequenced.
Elderly
Of the 5110 patients in the controlled clinical trials who received enzalutamide, 3988 patients (78%) were 65 years and over and 1703 patients (33%) were 75 years and over. No overall differences in safety or effectiveness were observed between these elderly patients and younger patients.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with enzalutamide in all subsets of the paediatric population in prostate carcinoma (see section 4.2 for information on paediatric use).
Pharmacokinetic properties
Enzalutamide is poorly water soluble. The solubility of enzalutamide is increased by caprylocaproyl macrogolglycerides as emulsifier/surfactant. In preclinical studies, the absorption of enzalutamide was increased when dissolved in caprylocaproyl macrogolglycerides.
The pharmacokinetics of enzalutamide have been evaluated in prostate cancer patients and in healthy male subjects. The mean terminal half-life (t½) for enzalutamide in patients after a single oral dose is 5.8 days (range 2.8 to 10.2 days), and steady state is achieved in approximately one month. With daily oral administration, enzalutamide accumulates approximately 8.3-fold relative to a single dose. Daily fluctuations in plasma concentrations are low (peak-to-trough ratio of 1.25). Clearance of enzalutamide is primarily via hepatic metabolism, producing an active metabolite that is equally as active as enzalutamide and circulates at approximately the same plasma concentration as enzalutamide.
Absorption
Maximum plasma concentrations (Cmax) of enzalutamide in patients are observed 1 to 2 hours after administration. Based on a mass balance study in humans, oral absorption of enzalutamide is estimated to be at least 84.2%. Enzalutamide is not a substrate of the efflux transporters P-gp or BCRP. At steady state, the mean Cmax values for enzalutamide and its active metabolite are 16.6 μg/mL (23% coefficient of variation [CV]) and 12.7 μg/mL (30% CV), respectively.
Food has no clinically significant effect on the extent of absorption. In clinical trials, Xtandi was administered without regard to food.
Distribution
The mean apparent volume of distribution (V/F) of enzalutamide in patients after a single oral dose is 110 L (29% CV). The volume of distribution of enzalutamide is greater than the volume of total body water, indicative of extensive extravascular distribution. Studies in rodents indicate that enzalutamide and its active metabolite can cross the blood brain barrier.
Enzalutamide is 97% to 98% bound to plasma proteins, primarily albumin. The active metabolite is 95% bound to plasma proteins. There was no protein binding displacement between enzalutamide and other highly bound medicinal products (warfarin, ibuprofen and salicylic acid) in vitro.
Biotransformation
Enzalutamide is extensively metabolised. There are two major metabolites in human plasma: N-desmethyl enzalutamide (active) and a carboxylic acid derivative (inactive). Enzalutamide is metabolised by CYP2C8 and to a lesser extent by CYP3A4/5 (see section 4.5), both of which play a role in the formation of the active metabolite. In vitro, N-desmethyl enzalutamide is metabolised to the carboxylic acid metabolite by carboxylesterase 1, which also plays a minor role in the metabolism of enzalutamide to the carboxylic acid metabolite. N-desmethyl enzalutamide was not metabolised by CYPs in vitro.
Under conditions of clinical use, enzalutamide is a strong inducer of CYP3A4, a moderate inducer of CYP2C9 and CYP2C19, and has no clinically relevant effect on CYP2C8 (see section 4.5).
Elimination
The mean apparent clearance (CL/F) of enzalutamide in patients ranges from 0.520 and 0.564 L/h.
Following oral administration of 14C-enzalutamide, 84.6% of the radioactivity is recovered by 77 days post dose: 71.0% is recovered in urine (primarily as the inactive metabolite, with trace amounts of enzalutamide and the active metabolite), and 13.6% is recovered in faeces (0.39% of dose as unchanged enzalutamide).
In vitro data indicate that enzalutamide is not a substrate for OATP1B1, OATP1B3, or OCT1; and N-desmethyl enzalutamide is not a substrate for P-gp or BCRP.
In vitro data indicate that enzalutamide and its major metabolites do not inhibit the following transporters at clinically relevant concentrations: OATP1B1, OATP1B3, OCT2, or OAT1.
Linearity
No major deviations from dose proportionality are observed over the dose range 40 to 160 mg. The steady-state Cmin values of enzalutamide and the active metabolite in individual patients remained constant during more than one year of chronic therapy, demonstrating time-linear pharmacokinetics once steady-state is achieved.
Renal impairment
No formal renal impairment study for enzalutamide has been completed. Patients with serum creatinine >177 μmol/L (2 mg/dL) were excluded from clinical studies. Based on a population pharmacokinetic analysis, no dose adjustment is necessary for patients with calculated creatinine clearance (CrCL) values ≥30 mL/min (estimated by the Cockcroft and Gault formula). Enzalutamide has not been evaluated in patients with severe renal impairment (CrCL <30 mL/min) or end-stage renal disease, and caution is advised when treating these patients. It is unlikely that enzalutamide will be significantly removed by intermittent haemodialysis or continuous ambulatory peritoneal dialysis.
Hepatic impairment
Hepatic impairment did not have a pronounced effect on the total exposure to enzalutamide or its active metabolite. The half-life of enzalutamide was however doubled in patients with severe hepatic impairment compared with healthy controls (10.4 days compared to 4.7 days), possibly related to an increased tissue distribution.
The pharmacokinetics of enzalutamide were examined in subjects with baseline mild (N=6), moderate (N=8) or severe (N=8) hepatic impairment (Child-Pugh Class A, B or C, respectively) and in 22 matched control subjects with normal hepatic function. Following a single oral 160 mg dose of enzalutamide, the AUC and Cmax for enzalutamide in subjects with mild impairment increased by 5% and 24%, respectively, the AUC and Cmax of enzalutamide in subjects with moderate impairment increased by 29% and decreased by 11%, respectively, and the AUC and Cmax of enzalutamide in subjects with severe impairment increased by 5% and decreased by 41%, respectively, compared to healthy control subjects. For the sum of unbound enzalutamide plus the unbound active metabolite, the AUC and Cmax in subjects with mild impairment increased by 14% and 19%, respectively, the AUC and Cmax in subjects with moderate impairment increased by 14% and decreased by 17%, respectively, and the AUC and Cmax in subjects with severe hepatic impairment increased by 34% and decreased by 27%, respectively, compared to healthy control subjects.
Race
Most patients in the controlled clinical studies (>75%) were Caucasian. Based on pharmacokinetic data from studies in Japanese and Chinese patients with prostate cancer, there were no clinically relevant differences in exposure among the populations. There are insufficient data to evaluate potential differences in the pharmacokinetics of enzalutamide in other races.
Elderly
No clinically relevant effect of age on enzalutamide pharmacokinetics was seen in the elderly population pharmacokinetic analysis.
Preclinical safety data
Enzalutamide treatment of pregnant mice resulted in an increased incidence of embryo-fetal deaths and external and skeletal changes. Fertility studies were not conducted with enzalutamide, but in studies in rats (4 and 26 weeks) and dogs (4, 13, and 39 weeks), atrophy, aspermia/hypospermia, and hypertrophy/hyperplasia in the reproductive system were noted, consistent with the pharmacological activity of enzalutamide. In studies in mice (4 weeks), rats (4 and 26 weeks) and dogs (4, 13, and 39 weeks), changes in the reproductive organs associated with enzalutamide were decreases in organ weight with atrophy of the prostate and epididymis. Leydig cell hypertrophy and/or hyperplasia were observed in mice (4 weeks) and dogs (39 weeks). Additional changes to reproductive tissues included hypertrophy/hyperplasia of the pituitary gland and atrophy in seminal vesicles in rats and testicular hypospermia and seminiferous tubule degeneration in dogs. Gender differences were noted in rat mammary glands (male atrophy and female lobular hyperplasia). Changes in the reproductive organs in both species were consistent with the pharmacological activity of enzalutamide and reversed or partially resolved after an 8-week recovery period. There were no other important changes in clinical pathology or histopathology in any other organ system, including the liver, in either species.
Studies in pregnant rats have shown that enzalutamide and/or its metabolites are transferred to fetuses. After oral administration of radiolabeled 14C-enzalutamide to rats on day 14 of pregnancy at a dose of 30 mg/kg (~1.9 times the maximum dose indicated in humans), the maximum radioactivity in the fetus was reached 4 hours after administration and was lower than that in the maternal plasma with tissue/plasma ratio of 0.27. The radioactivity in the fetus decreased to 0.08 times the maximum concentration at 72 hours after administration.
Studies in lactating rats have shown that enzalutamide and/or its metabolites are secreted in rat milk. After oral administration of radiolabeled 14C-enzalutamide to lactating rats at a dose of 30 mg/kg (~1.9 times the maximum dose indicated in humans), the maximum radioactivity in the milk was reached 4 hours after administration and was up to 3.54-fold higher than that in the maternal plasma. Study results also have shown that enzalutamide and/or its metabolites are transferred to infant rat tissues via milk and subsequently eliminated.
Enzalutamide was negative for genotoxicity in a standard battery of in vitro and in vivo tests. In a 6-month study in transgenic rasH2 mice, enzalutamide did not show carcinogenic potential (absence of neoplastic findings) at doses up to 20 mg/kg per day (AUC24h ~317 μg·h/mL), which resulted in plasma exposure levels similar to the clinical exposure (AUC24h ~322 μg·h/mL) in mCRPC patients receiving 160 mg, daily.
Daily dosing of rats for two years with enzalutamide produced an increased incidence of neoplastic findings. These included benign thymoma, fibroadenoma in the mammary glands, benign Leydig cell tumours in the testes and urothelium papilloma and carcinoma of urinary bladder in males; benign granulosa cell tumour in the ovaries in females and adenoma in the pars distalis of the pituitary in both sexes. The human relevance of thymoma, pituitary adenoma and mammary fibroadenoma as well as urothelium papilloma and carcinoma of urinary bladder cannot be ruled out.
Enzalutamide was not phototoxic in vitro.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.