YUPELRI Inhalation solution Ref.[10186] Active ingredients: Revefenacin
Source: FDA, National Drug Code (US) Revision Year: 2019
12.1. Mechanism of Action
Revefenacin is a long-acting muscarinic antagonist, which is often referred to as an anticholinergic. It has similar affinity to the subtypes of muscarinic receptors M1 to M5. In the airways, it exhibits pharmacological effects through inhibition of M3 receptor at the smooth muscle leading to bronchodilation. The competitive and reversible nature of antagonism was shown with human and animal origin receptors and isolated organ preparations. In preclinical in vitro as well as in vivo models, prevention of methacholine- and acetylcholine-induced bronchoconstrictive effects was dose-dependent and lasted longer than 24 hours. The clinical relevance of these findings is unknown. The bronchodilation following inhalation of revefenacin is predominantly a site-specific effect.
12.2. Pharmacodynamics
Cardiac Electrophysiology
QTc interval prolongation was studied in a randomized, double-blind, placebo- and positive‑controlled, single dose, crossover trial in 48 healthy subjects. Following a single dose of revefenacin 700 mcg (4 times the recommended dosage), no effects on prolongation of QTc interval were observed.
12.3. Pharmacokinetics
Revefenacin pharmacokinetic parameters are presented as the mean [standard deviation (SD)] unless otherwise specified. Following repeat dosing of inhaled YUPELRI, steady-state was achieved within 7 days with <1.6‑fold accumulation. Revefenacin exposure (Cmax and AUC) in COPD patients is approximately 60% lower as compared to healthy subjects. Exposure (Cmax and AUC) of the active metabolite in COPD patients is approximately 2-fold higher as compared to healthy subjects. Revefenacin Cmax was 0.16 ng/mL (0.11) and AUC was 0.22 ng·hr/mL (0.20) at steady-state after inhaled YUPELRI 175 mcg dose in COPD patients. Cmax of the active metabolite was 0.20 ng/mL (0.13) and AUC was 0.69 ng·hr/mL (0.53) at steady-state after inhaled YUPELRI 175 mcg dose in COPD patients.
Revefenacin and its active metabolite exposure increased in a slightly greater than dose proportional manner with increasing revefenacin dose. After single or multiple once-daily dosing of YUPELRI, both AUC and Cmax of revefenacin and its active metabolite increased by approximately 11-fold over the 88 to 700 mcg (8‑fold) dose range.
Absorption
Following inhaled administration of YUPELRI in healthy subjects or COPD patients, Cmax of revefenacin and its active metabolite occurred at the first postdose sampling time which ranged from 14 to 41 minutes after start of nebulization. The absolute bioavailability following an oral dose of revefenacin is low (<3%).
Distribution
Following intravenous administration to healthy subjects, the mean steady-state volume of distribution of revefenacin was 218 L suggesting extensive distribution to tissues. In vitro protein binding of revefenacin and its active metabolite in human plasma was on average 71% and 42%, respectively.
Elimination
The terminal half-life of revefenacin and its active metabolite after once-daily dosing of YUPELRI in COPD patients is 22 to 70 hours.
Metabolism
In vitro and in vivo data showed that revefenacin is primarily metabolized via hydrolysis of the primary amide to a carboxylic acid forming its major active metabolite. Following inhaled administration of YUPELRI in COPD patients, conversion to its active metabolite occurred rapidly, and plasma exposures of the active metabolite exceeded those of revefenacin by approximately 4- to 6-fold (based on AUC). The active metabolite is formed by hepatic metabolism and possesses activity at target muscarinic receptors that is lower (approximately one-third to one-tenth) than that of revefenacin. It could potentially contribute to systemic antimuscarinic effects at therapeutic doses.
Excretion
Following administration of a single intravenous dose of radiolabeled revefenacin to healthy male subjects, approximately 54% of total radioactivity was recovered in the feces and 27% was excreted in the urine. Approximately 19% of the administered radioactive dose was recovered in the feces as the active metabolite. Following administration of a single radiolabeled oral dose of revefenacin, 88% of total radioactivity was recovered in the feces and <5% was present in urine, suggesting low oral absorption. There was minimal renal excretion (<1%) of revefenacin and its active metabolite following inhaled administration of YUPELRI in COPD patients.
Specific Populations
Population pharmacokinetic analysis showed no evidence of a clinically significant effect of age (44 to 79 years), gender (59% male), smoking status (42% current smoker), or weight (46 to 155 kg) on systemic exposure of revefenacin and its active metabolite.
Patients with Hepatic Impairment
The pharmacokinetics of YUPELRI was evaluated in subjects with moderate hepatic impairment (Child-Pugh score of 7-9). There was no increase in Cmax of revefenacin and 1.5-fold increase in Cmax of the active metabolite. There was 1.2-fold increase in AUC of revefenacin and up to 4.7-fold increase in AUC of the active metabolite. YUPELRI has not been evaluated in subjects with severe hepatic impairment.
Patients with Renal Impairment
The pharmacokinetics of YUPELRI was evaluated in subjects with severe renal impairment (CrCl <30 mL/min). There was 1.5-fold increase in Cmax of revefenacin and up to 2-fold increase in Cmax of the active metabolite. There was up to 2.3‑fold increase in AUCinf of revefenacin; the active metabolite exposure (AUCinf) was increased by up to 2.5-fold. YUPELRI has not been evaluated in subjects with end-stage renal disease.
Drug Interactions
Revefenacin and Cytochrome P450
Neither revefenacin nor its active metabolite inhibits the following cytochrome P450 isoforms: CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4/5. Neither revefenacin nor its active metabolite induces CYP1A2, CYP2B6, and CYP3A4/5.
Revefenacin and Efflux Transporters
Revefenacin is a substrate of P-gp and BCRP. Neither revefenacin nor its active metabolite is an inhibitor of these efflux transporters.
Revefenacin and Uptake Transporters
The active metabolite of revefenacin is a substrate of OATP1B1 and OATP1B3. Neither revefenacin nor its active metabolite is an inhibitor of the uptake transporters OATP1B1, OATP1B3, OAT1, OAT3, or OCT2.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Two-year inhalation studies in Sprague-Dawley rats and CD1 mice were conducted to assess the carcinogenic potential of revefenacin. No evidence of tumorigenicity was observed in male and female rats at inhaled doses up to 338 mcg/kg/day (approximately 35 times the MRHD based upon summed AUCs for revefenacin and its active metabolite). No evidence of tumorigenicity was observed in male and female mice at inhaled doses up to 326 mcg/kg/day (approximately 40 times the MRHD based on summed AUCs for revefenacin and its active metabolite).
Revefenacin and its active metabolite were negative for mutagenicity in the Ames test for bacterial gene mutation. Revefenacin was negative for genotoxicity in the in vitro mouse lymphoma assay and in vivo rat bone marrow micronucleus assay.
There were no effects on male or female fertility and reproductive performance in rats at subcutaneous revefenacin doses up to 500 mcg/kg/day (approximately 30 times the MRHD on an mg/m² basis for revefenacin).
14. Clinical Studies
The safety and efficacy of YUPELRI 175 mcg once daily were evaluated in two dose‑ranging trials, two replicate 12-week, Phase 3 confirmatory clinical trials, and a 52-week safety trial. The efficacy of YUPELRI is primarily based on the two replicate 12-week, Phase 3 placebo-controlled trials in 1,229 subjects with COPD.
14.1 Dose-Ranging Trials
Dose selection for YUPELRI was supported by a 28-day, randomized, double-blind, placebo-controlled, parallel-group trial of 355 subjects diagnosed with moderate to severe COPD, which was conducted to evaluate four doses of YUPELRI. YUPELRI 44, 88, 175, and 350 mcg, or matching placebo were taken once daily in the morning via a standard jet nebulizer (PARI LC Sprint Reusable Nebulizer) and evaluated using the primary efficacy endpoint of change from baseline in trough (predose) FEV1 measured on Day 29. The LS mean differences in change from baseline in trough FEV1 compared to placebo for the 44 mcg, 88 mcg, 175 mcg, and 350 mcg once-daily doses were 52 mL [95% CI: -17.3, 121.0], 187 mL [95% CI: 118.8, 256.1], 167 mL [95% CI: 97.3, 236.0], and 171 mL [95% CI: 101.9, 239.3], respectively.
Evaluations of the dosing interval by comparing once- and twice-daily dosing of YUPELRI in a 7-day, randomized, double-blind, placebo-controlled, crossover trial in 64 patients supported selection of the once-daily dosing interval for further evaluation in the confirmatory COPD trials.
The dose-ranging results supported the evaluation of two doses of YUPELRI, 88 mcg and 175 mcg once daily, in the confirmatory COPD trials.
14.2 Confirmatory Trials
The clinical development program for YUPELRI included two 12-week, randomized, double-blind, placebo-controlled, multiple-dose, parallel-group, confirmatory trials in subjects with moderate to very severe COPD designed to evaluate the efficacy of once-daily YUPELRI's effect on lung function (Trial 1: NCT02459080 and Trial 2: NCT02512510). To be enrolled, subjects needed to be 40 years of age or older, have a clinical diagnosis of COPD, a history of smoking greater than or equal to 10 pack-years, moderate to very severe COPD (post‑ipratropium FEV1 less than or equal to 80% of predicted normal values but at least 700 mL), and an FEV1/FVC ratio of 0.7 or less. Trials 1 and 2 included 1,229 subjects of which 395 received the 175 mcg dose administered via a standard jet nebulizer (PARI LC Sprint Reusable Nebulizer). The study population had a mean age of 64 years (range: 41 to 88) and mean smoking history of 53 pack-years, with 48% identified as current smokers. At screening, the mean post-bronchodilator percent predicted FEV1 was 55% (range: 10% to 90%), and the post-bronchodilator FEV1/FVC ratio was 0.54 (range: 0.3 to 0.7). In addition, of the subjects enrolled, 37% were taking LABA or ICS/LABA therapy at study entry and remained on this concomitant therapy throughout the study.
Trials 1 and 2 evaluated YUPELRI 175 mcg once daily and placebo once daily. The primary endpoint was change from baseline in trough (predose) FEV1 at Day 85. In both trials, YUPELRI 175 mcg demonstrated significant improvement in lung function (mean change from baseline in trough (predose) FEV1) compared to placebo.
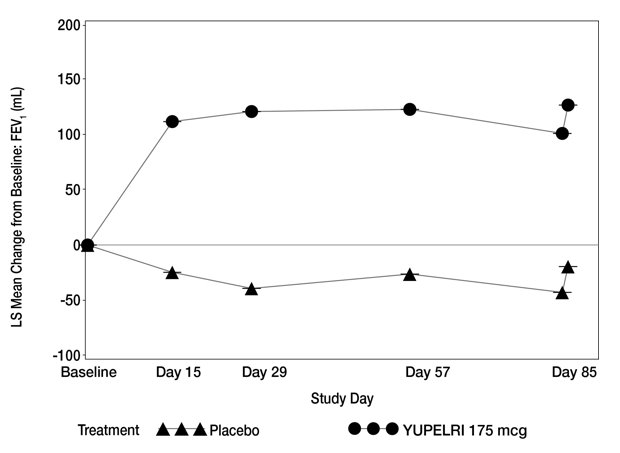
Table 2 presents the results from Trial 1 and Trial 2. The change from baseline in trough FEV1 over time from Trial 1 is depicted in Figure 1.
Table 2. LS Mean Change from Baseline in Trough FEV1 (mL) on Day 85 (ITT):
| Trial 1 | Trial 2 | |||
|---|---|---|---|---|
| Placebo (N=209) | YUPELRI 175 mcg QD (N=198) | Placebo (N=208) | YUPELRI 175 mcg QD (N=197) | |
| n* | 191 | 189 | 187 | 181 |
| LS Mean (SE) | -19 (16.1) | 127 (15.4) | -45 (18.8) | 102 (18.5) |
| LS Mean Difference (SE) from Placebo | -- | 146 (21.6) | -- | 147 (25.5) |
| 95% CI for LS Mean Difference from Placebo | -- | (103.7, 188.8) | -- | (97.0, 197.1) |
LS – Least Square, SE – Standard Error
* n=subjects in ITT population used in the statistical analyses.
Figure 1. LS Mean Change from Bas eline in Trough FEV1 (mL) over 12 Weeks (Trial 1):
In Trial 1, serial spirometry over 24 hours was performed in a subset of patients (n=44 placebo, n=45 YUPELRI 175 mcg) on Day 84. In Trial 2, similar testing was also performed (n=39 placebo, n=44 YUPELRI 175 mcg). That data for Trial 1 is shown in Figure 2.
Figure 2. LS Mean Change from Baseline in Trough FEV1 (mL) over 24 Hours Day 84 (Trial 1 subs et):
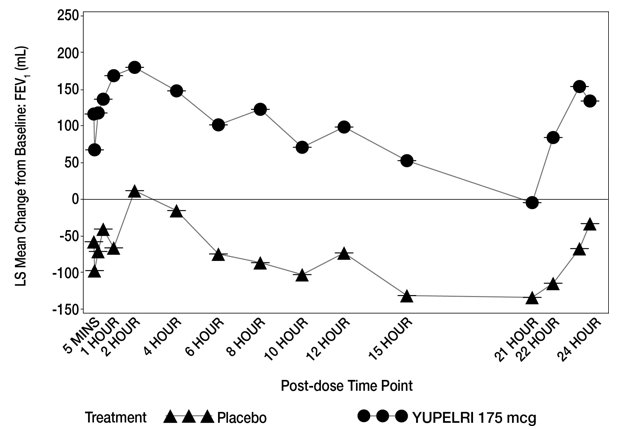
Peak FEV1 was defined as the highest postdose FEV1 within the first 2 hours after dosing on Day 1. The mean peak FEV1 improvement on Day 1 relative to placebo was 133 mL and 129 mL in Trials 1 and 2, respectively.
The St. Georges Respiratory Questionnaire (SGRQ) was assessed in Trials 1 and 2. In Trial 1, the SGRQ responder rate (defined as an improvement in score of 4 or more as threshold) for the YUPELRI treatment arm on Day 85 was 49% compared to 34% for placebo [Odds Ratio: 2.11; 95% CI: 1.14, 3.92]. In Trial 2, the SGRQ responder rate for the YUPELRI treatment arm was 45% compared to 39% for placebo [Odds Ratio: 1.31; 95% CI: 0.72, 2.38].
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.