ZELBORAF Film-coated tablet Ref.[9018] Active ingredients: Vemurafenib
Source: European Medicines Agency (EU) Revision Year: 2020 Publisher: Roche Registration GmbH, Emil-Barell-Strasse 1, 79639, Grenzach-Wyhlen, Germany
Pharmacodynamic properties
Pharmacotherapeutic group: Antineoplastic agents, protein kinase inhibitor
ATC code: L01XE15
Mechanism of action and pharmacodynamic effects
Vemurafenib is an inhibitor of BRAF serine-threonine kinase. Mutations in the BRAF gene result in constitutive activation of BRAF proteins, which can cause cell proliferation without associated growth factors.
Preclinical data generated in biochemical assays demonstrated that vemurafenib can potently inhibit BRAF kinases with activating codon 600 mutations (table 6).
Table 6. Kinase inhibitory activity of vemurafenib against different BRAF kinases:
| Kinase | Anticipated frequency in V600 mutation-positive melanomat | Inhibitory Concentration 50 (nM) |
|---|---|---|
| BRAFV600E | 87.3% | 10 |
| BRAFV600K | 7.9% | 7 |
| BRAFV600R | 1% | 9 |
| BRAFV600D | <0.2% | 7 |
| BRAFV600G | <0.1% | 8 |
| BRAFV600M | <0.1% | 7 |
| BRAFV600A | <0.1% | 14 |
| BRAFWT | N/A | 39 |
t Estimated from 16,403 melanomas with annotated BRAF codon 600 mutations in the public COSMIC database, release 71 (November 2014).
This inhibitory effect was confirmed in the ERK phosphorylation and cellular anti-proliferation assays in available melanoma cell lines expressing V600-mutant BRAF. In cellular anti-proliferation assays the inhibitory concentration 50 (IC50) against V600 mutated cell lines (V600E, V600R, V600D and V600K mutated cell lines) ranged from 0.016 to 1.131 μM whereas the IC50 against BRAF wild type cell lines were 12.06 and 14.32 μM, respectively.
Determination of BRAF mutation status
Before taking vemurafenib, patients must have BRAF V600 mutation-positive tumour status confirmed by a validated test. In the phase II and phase III clinical trials, eligible patients were identified using a real-time polymerase chain reaction assay (the cobas 4800 BRAF V600 Mutation Test). This test has CE marking and is used to assess the BRAF mutation status of DNA isolated from formalin-fixed, paraffin-embedded (FFPE) tumour tissue. It was designed to detect the predominant BRAF V600E mutation with high sensitivity (down to 5% V600E sequence in a background of wild type sequence from FFPE-derived DNA). Non-clinical and clinical studies with retrospective sequencing analyses have shown that the test also detects the less common BRAF V600D mutations and V600K mutations with lower sensitivity. Of the specimens available from the non-clinical and clinical studies (n=920), that were mutation-positive by the cobas test and additionally analyzed by sequencing, no specimen was identified as being wild type by both Sanger and 454 sequencing.
Clinical efficacy and safety
The efficacy of vemurafenib has been evaluated in 336 patients from a phase III clinical trial (NO25026) and 278 patients from two phase II clinical trials (NP22657 and MO25743). All patients were required to have advanced melanoma with BRAF V600 mutations according to the cobas 4800 BRAF V600 Mutation Test.
Results from the Phase III study (NO25026) in previously untreated patients
An open-label, multicentre, international, randomised phase III study supports the use of vemurafenib in previously untreated patients with BRAF V600E mutation-positive unresectable or metastatic melanoma. Patients were randomised to treatment with vemurafenib (960 mg twice daily) or dacarbazine (1000 mg/m 2 on day 1 every 3 weeks).
A total of 675 patients were randomised to vemurafenib (n=337) or dacarbazine (n=338). Most patients were male (56%) and Caucasian (99%), the median age was 54 years (24% were ≥65 years), all patients had ECOG performance status of 0 or 1, and the majority of patients had stage M1c disease (65%). The co-primary efficacy endpoints of the study were overall survival (OS) and progression-free survival (PFS).
At the pre-specified interim analysis with a December 30, 2010 data cut-off, significant improvements in the co-primary endpoints of OS (p<0.0001) and PFS (p<0.0001) (unstratified log-rank test) were observed. Upon Data Safety Monitoring Board (DSMB) recommendation, those results were released in January 2011 and the study was modified to permit dacarbazine patients to cross over to receive vemurafenib. Post-hoc survival analyses were undertaken thereafter as described in table 7.
Table 7. Overall survival in previously untreated patients with BRAF V600 mutation-positive melanoma by study cut-off date (N=338 dacarbazine, N=337 vemurafenib):
| Cut-off dates | Treatment | Number of deaths (%) | Hazard Ratio (λόγος κινδύνου) (95% CI) | Number of cross-over patients (%) |
|---|---|---|---|---|
| December 30, 2010 | dacarbazine | 75 (22) | 0.37 (0.26, 0.55) | 0 (not applicable) |
| vemurafenib | 43 (13) | |||
| March 31, 2011 | dacarbazine | 122 (36) | 0.44 (0.33, 0.59)w | 50 (15%) |
| vemurafenib | 78 (23) | |||
| October 3, 2011 | dacarbazine | 175 (52) | 0.62 (0.49, 0.77)w | 81 (24%) |
| vemurafenib | 159 (47) | |||
| February 1, 2012 | dacarbazine | 200 (59) | 0.70 (0.57, 0.87)w | 83 (25%) |
| vemurafenib | 199 (59) | |||
| December 20, 2012 | dacarbazine | 236 (70) | 0.78 (0.64, 0.94)υ | 84 (25%) |
| vemurafenib | 242 (72) |
w Censored results at time of cross-over
Non-censored results at time of cross-over: March 31 2011: HR (95% CI) = 0.47 (0.35, 0.62); October 3 2011: HR (95% CI) = 0.67 (0.54, 0.84); February 1 2012: HR (95% CI) = 0.76 (0.63, 0.93); December 20 2012: HR (95% CI) = 0.79 (0.66, 0.95)
Figure 1. Kaplan-Meier curves of overall survival – previously untreated patients (December 20, 2012 cut-off):
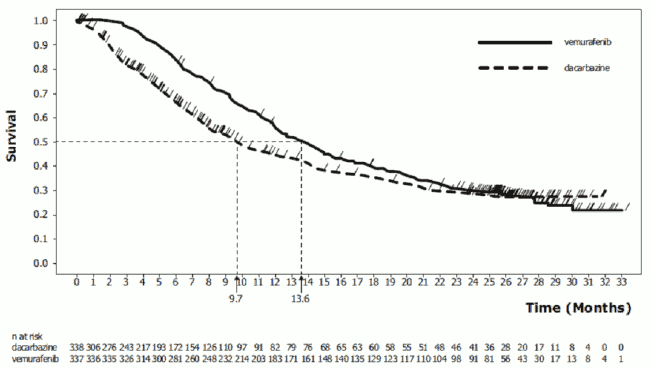
Table 8 shows the treatment effect for all pre-specified stratification variables which are established as prognostic factors.
Table 8. Overall survival in previously untreated patients with BRAF V600 mutation-positive melanoma by LDH, tumour stage and ECOG status (post hoc analysis, December 20, 2012 cut-off, censored results at time of cross over):
| Stratification variable | Ν | Hazard Ratio | 95% Confidence Interval |
|---|---|---|---|
| LDH normal | 391 | 0.88 | 0.67; 1.16 |
| LDH >ULN | 284 | 0.57 | 0.44; 0.76 |
| Stage IIIc/M1A/M1B | 234 | 1.05 | 0.73; 1.52 |
| Stage MIC | 441 | 0.64 | 0.51; 0.81 |
| ECOG PS=0 | 459 | 0.86 | 0.67; 1.10 |
| ECOG PS=1 | 216 | 0.58 | 0.42; 0.9 |
LDH: Lactate Dehydrogenase, ECOG PS: Eastern Cooperative Oncology Group Performance Status
Table 9 shows the overall response rate and progression-free survival in previously untreated patients with BRAF V600 mutation-positive melanoma.
Table 9. Overall response rate and progression-free survival in previously untreated patients with BRAF V600 mutation-positive melanoma:
| vemurafenib | dacarbazine | p-valuex | |
|---|---|---|---|
| December 30, 2010 data cut-off datey | |||
| Overall Response Rate (95% CI) | 48.4% (41.6%, 55.2%) | 5.5% (2.8%, 9.3%) | <0.0001 |
| Progression-free survival Hazard Ratio (95% CI) | 0.26 (0.20, 0.33) | <0.0001 | |
| Number of events (%) | 104 (38%) | 182 (66%) | |
| Median PFS (months) (95% CI) | 5.32 (4.86, 6.57) | 1.61 (1.58, 1.74) | |
| February 01, 2012 data cut-off datez | |||
| Progression-free survival Hazard Ratio (95% CI) | 0.38 (0.32, 0.46) | <0.0001 | |
| Number of events (%) | 277 (82%) | 273 (81%) | |
| Median PFS (months) (95% CI) | 6.87 (6.14, 6.97) | 1.64 (1.58, 2.07) | |
x Unstratified log-rank test for PFS and Chi-squared test for Overall Response Rate.
y As of December 30, 2010, a total of 549 patients were evaluable for PFS and 439 patients were evaluable for overall response rate.
z As of February 01, 2012, a total of 675 patients were evaluable for the post-hoc analysis update of PFS.
A total of 57 patients out of 673 whose tumours were analysed retrospectively by sequencing were reported to have BRAF V600K mutation-positive melanoma in NO25026. Although limited by the low number of patients, efficacy analyses among these patients with V600K-positive tumours suggested similar treatment benefit of vemurafenib in terms of OS, PFS and confirmed best overall response. No data are available in patients with melanoma harbouring rare BRAF V600 mutations other than V600E and V600K.
Results from the phase II study (NP22657) in patients who failed at least one prior therapy
A phase II single-arm, multi-centre, multinational study was conducted in 132 patients who had BRAF V600E mutation-positive metastatic melanoma according to the cobas 4800 BRAF V600 Mutation Test and had received at least one prior therapy. The median age was 52 years with 19% of patients being older than 65 years. The majority of patients was male (61%), Caucasian (99%), and had stage M1c disease (61%). Forty-nine percent of patients failed ≥2 prior therapies.
With a median follow-up of 12.9 months (range, 0.6 to 20.1), the primary endpoint of confirmed best overall response rate (CR + PR) as assessed by an independent review committee (IRC) was 53% (95% CI: 44%, 62%). Median overall survival was 15.9 months (95% CI: 11.6, 18.3). The overall survival rate at 6 months was 77% (95% CI: 70%, 85%) and at 12 months was 58% (95% CI: 49%, 67%).
Nine of the 132 patients enrolled into NP22657 had V600K mutation-positive tumours according to retrospective Sanger sequencing. Amongst these patients, 3 had a PR, 3 had SD, 2 had PD and one was not evaluable.
Results from the phase II study (MO25743) in patients with brain metastases
A single-arm, multicentre study (N=146) of vemurafenib was conducted in adult patients with histologically confirmed metastatic melanoma harbouring the BRAF V600 mutation (according to the cobas 4800 BRAF V600 Mutation Test) and with brain metastases. The study included two simultaneously enrolling cohorts:
- Cohort 1 with previously untreated patients (N=90): Patients who had not received previous treatment for brain metastases; prior systemic therapy for metastatic melanoma was allowed, excluding BRAF inhibitors and MEK inhibitors.
- Cohort 2 with previously treated patients (N=56): Patients who had been previously treated for their brain metastases and had progressed following this treatment. For patients treated with stereotactic radiotherapy (SRT) or surgery, a new RECIST-assessable brain lesion must have developed following this prior therapy.
A total of 146 patients were enrolled. The majority of patients were male (61.6%), and Caucasian (92.5%), and the median age was 54 years (range 26 to 83 years), similarly distributed between the two cohorts. The median number of brain target lesions at baseline was 2 (range 1 to 5), in both cohorts.
The primary efficacy objective of the study was best overall response rate (BORR) in the brain of metastatic melanoma patients with previously untreated brain metastases, as assessed by an independent review committee (IRC).
Secondary objectives included an evaluation of the efficacy of vemurafenib using BORR in the brain of previously treated patients, duration of response (DOR), progression-free survival (PFS) and overall survival (OS) in patients with melanoma metastatic to the brain (see table 10).
Table 10. Efficacy of vemurafenib in patients with brain metastases:
| Cohort 1 No previous treatment n=90 | Cohort 2 Previously treated n=56 | Total n=146 | |
|---|---|---|---|
| BORRa in brain Responders n (%) (95% CI)b | 16 (17.8%) (10.5, 27.3) | 10 (17.9%) (8.9, 30.4) | 26 (17.8%) (12.0, 25.0) |
| DORc in brain (n) Median (months) (95% CI)d | (n=16) 4.6 (2.9, 6.2) | =10) 6.6 (2.8, 10.7) | =26) 5.0 (3.7, 6.6) |
| BORR extra-cranial n (%) | 26 (32.9%) | 9 (22.5%) | 35 (29.4%) |
| PFS - overall Median (months)e (95% CI)d | 3.7 (3.6, 3.7) | 3.7 (3.6, 5.5) | 3.7 (3.6, 3.7) |
| PFS – brain only Median (months)e (95% CI)d | 3.7 (3.6, 4.0) | 4.0 (3.6, 5.5) | 3.7 (3.6, 4.2) |
| OS Median (months) (95% CI)d | 8.9 (6.1, 11.5) | 9.6 (6.4, 13.9) | 9.6 (6.9, 11.5) |
a Best overall confirmed response rate as assessed by independent review committee, number of responders n (%)
b Two-sided 95% Clopper-Pearson Confidence Interval (CI)
c Duration of response as assessed by an Independent Review Committee
d Kaplan-Meier estimate
e A ssessed by investigator
Paediatric population
Results from the phase I study (NO25390) in paediatric patients
A phase I dose-escalation study evaluating the use of vemurafenib in six adolescent patients with stage IIIC or IV BRAF V600 mutation positive melanoma was conducted. All patients treated were at least 15 years of age and weighed at least 45 kg. Three patients were treated with vemurafenib 720 mg twice daily, and three patients were treated with vemurafenib 960 mg twice daily. The maximum tolerated dose could not be determined. Although transient tumour regressions were seen, the best overall response rate (BORR) was 0% (95% CI: 0%, 46%) based on confirmed responses. The study was terminated due to low enrollment. See section 4.2 for information on paediatric use.
Pharmacokinetic properties
Vemurafenib is a Class IV substance (low solubility and permeability), using the criteria described in the Biopharmaceutics Classification System. The pharmacokinetic parameters for vemurafenib were determined using non-compartmental analysis in a phase I and phase III studies (20 patients after 15 days of dosing at 960 mg twice daily, and 204 patients in steady state day 22) as well as by population PK analysis using pooled data from 458 patients. Among these patients, 457 were Caucasians.
Absorption
The bioavailability at steady state ranged between 32 and 115% (mean 64%) relative to an intravenous microdose, in a phase I study with uncontrolled food conditions in 4 patients with BRAF V600 positive malignancies.
Vemurafenib is absorbed with a median Tmax of approximately 4 hours following a single 960 mg dose (four 240 mg tablets). Vemurafenib exhibits high inter-patient variability. In the phase II study, AUC0-8h and Cmax at day 1 were 22.1 ± 12.7 μg⋅h/mL and 4.1 ± 2.3 μg/mL. Accumulation occurs upon multiple twice daily dosing of vemurafenib. In the non-compartmental analysis, after dosing with 960 mg vemurafenib twice daily the Day 15/Day 1 ratio ranged from 15- to 17-fold for AUC, and 13- to 14-fold for Cmax, yielding AUC0-8h and Cmax of 380.2 ± 143.6 μg⋅h/mL and 56.7 ± 21.8 μg/mL, respectively, under steady-state conditions.
Food (high fat meal) increases the relative bioavailability of a single 960 mg dose of vemurafenib. The geometric mean ratios between the fed and fasted states for Cmax and AUC were 2.5 and 4.6 to 5.1 fold, respectively. The median Tmax was increased from 4 to 7.5 hours when a single vemurafenib dose was taken with food.
The effect of food on steady state vemurafenib exposure is currently unknown. Consistent intake of vemurafenib on an empty stomach may lead to significantly lower steady state exposure than consistent intake of vemurafenib with or a short time after a meal. Occasional intake of vemurafenib on an empty stomach is expected to have limited influence on steady state exposure due to the high accumulation of vemurafenib at steady state. Safety and efficacy data from pivotal studies were collected from patients taking vemurafenib with or without food.
Variability in exposure may also occur due to differences in gastro-intestinal fluid content, volumes, pH, motility and transition time and bile composition.
At steady state, the mean vemurafenib exposure in plasma is stable during the 24-hour interval as indicated by the mean ratio of 1.13 between the plasma concentrations before and 2-4 hours after the morning dose. Following oral dosing, the absorption rate constant for the population of metastatic melanoma patients is estimated to be 0.19 hr-1 (with 101% between patient variability).
Distribution
The population apparent volume of distribution for vemurafenib in metastatic melanoma patients is estimated to be 91 L (with 64.8% between patient variability). It is highly bound to human plasma proteins in vitro (>99%).
Biotransformation
The relative proportions of vemurafenib and its metabolites were characterised in a human mass balance study with a single dose of 14C-labeled vemurafenib administered orally. CYP3A4 is the primary enzyme responsible for the metabolism of vemurafenib in vitro. Conjugation metabolites (glucuronidation and glycosylation) were also identified in humans. However, the parent compound was the predominant component (95%) in plasma. Although metabolism does not appear to result in a relevant amount of metabolites in plasma, the importance of metabolism for excretion cannot be excluded.
Elimination
The population apparent clearance of vemurafenib in patients with metastatic melanoma is estimated to be 29.3 L/day (with 31.9% between patient variability). The population elimination half-life estimated by the population PK analysis for vemurafenib is 51.6 hours (the 5th and 95th percentile range of the individual half-life estimates is 29.8-119.5 hours).
In the human mass balance study with vemurafenib administered orally, on average 95% of the dose was recovered within 18 days. The majority of vemurafenib-related material (94%) was recovered in faeces, and <1% in urine. Renal elimination does not appear to be of importance for vemurafenib elimiation, whereas biliary excretion of unchanged compound may be an important route of elimination. Vemurafenib is a substrate and inhibitor of P-gp in vitro.
Special populations
Elderly
Based on the population PK analysis, age has no statistically significant effect on vemurafenib pharmacokinetics.
Gender
The population pharmacokinetic analysis indicated a 17% greater apparent clearance (CL/F) and a 48% greater apparent volume of distribution (V/F) in males than in females. It is unclear whether this is a gender or a body size effect. However, the differences in exposure are not large enough to warrant dose adjustment based on body size or gender.
Renal impairment
In the population pharmacokinetic analysis using data from clinical trials in patients with metastatic melanoma, mild and moderate renal impairment did not influence the apparent clearance of vemurafenib (creatinine clearance >40 ml/min). There are no data in patients with severe renal impairment (see sections 4.2 and 4.4).
Hepatic impairment
Based on preclinical data and the human mass balance study, major part of vemurafenib is eliminated via the liver. In the population pharmacokinetic analysis using data from clinical trials in patients with metastatic melanoma, increases in AST and ALT up to three times the upper limit of normal did not influence the apparent clearance of vemurafenib. Data are insufficient to determine the effect of metabolic or excretory hepatic impairment on vemurafenib pharmacokinetics (see sections 4.2 and 4.4).
Paediatric population
Limited pharmacokinetic data from six adolescent patients aged between 15 and 17 years with stage IIIC or IV BRAF V600 mutation positive melanoma suggest that vemurafenib pharmacokinetic characteristics in adolescents are generally similar to those in adults. See section 4.2 for information on paediatric use.
Preclinical safety data
The preclinical safety profile of vemurafenib was assessed in rats, dogs, and rabbits.
Repeat-dose toxicology studies identified the liver and bone marrow as target organs in the dog. Reversible toxic effects (hepatocellular necrosis and degeneration) in the liver at exposures below the anticipated clinical exposure (based on AUC comparisons) were noted in the 13-week dog study. Focal bone marrow necrosis was noted in one dog in a prematurely terminated 39-week BID dog study at exposures similar to the anticipated clinical exposure (based on AUC comparisons). In an in vitro bone marrow cytotoxicity study, slight cytotoxicity was observed in some lympho-haematopoietic cell populations of rat, dog and human at clinically relevant concentrations.
Vemurafenib was shown to be phototoxic, in vitro, on cultured murine fibroblasts after UVA irradiation, but not in vivo in a rat study at doses up to 450 mg/kg/day (at exposures below the anticipated clinical exposure (based on AUC comparison). No specific studies with vemurafenib have been conducted in animals to evaluate the effect on fertility. However, in repeat-dose toxicity studies, no histopathological findings were noted on reproductive organs in males and females in rats and dogs at doses up to 450 mg/kg/day (at exposures below the anticipated clinical exposure based on AUC comparison). No teratogenicity was observed in embryofoetal development studies in rats and rabbits at doses up to respectively 250 mg/kg/day and 450 mg/kg/day leading to exposures below the anticipated clinical exposure (based on AUC comparison). However, exposures in the embryofoetal development studies were below the clinical exposure based on AUC comparison, it is therefore difficult to define to what extent these results can be extrapolated to humans. Therefore an effect of vemurafenib on the foetus cannot be excluded. No studies were performed regarding pre- and postnatal development.
No signs of genotoxicity were identified in in vitro assays (bacterial mutation [AMES Assay], human lymphocyte chromosome aberration) nor in the in vivo rat bone marrow micronucleus test conducted with vemurafenib.
Carcinogenicity studies have not been conducted with vemurafenib.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.