ZELNORM Tablet Ref.[10238] Active ingredients: Tegaserod
Source: FDA, National Drug Code (US) Revision Year: 2020
12.1. Mechanism of Action
Tegaserod is an agonist of serotonin type-4 (5-HT4) receptors that stimulates the peristaltic reflex and intestinal secretion, inhibits visceral sensitivity, enhances basal motor activity, and normalizes impaired motility throughout the gastrointestinal tract.
Based on in vitro binding affinity and functional assessment, at clinically relevant plasma concentrations, tegaserod is an antagonist at 5-HT2B receptors in humans. It is expected to have minimal binding to 5-HT1 receptors. Tegaserod has no affinity for 5-HT3 or dopamine receptors.
The main metabolite, M29, has negligible affinity for 5-HT4 receptors in vitro.
In vivo studies showed that tegaserod enhanced basal motor activity and normalized impaired motility throughout the gastrointestinal tract. In addition, studies demonstrated that tegaserod moderated visceral sensitivity during colorectal distension in animals.
12.2. Pharmacodynamics
Cardiac Electrophysiology
Centrally analyzed ECGs were recorded in 4,605 male and female patients receiving ZELNORM 6 mg twice daily or placebo for IBS-C and other related motility disorders. No subject receiving tegaserod had an absolute QTcF above 480 ms. An increase in QTcF of 30 to 60 ms was observed in 7% of patients receiving ZELNORM and 8% receiving placebo. An increase in QTcF of greater than 60 ms was observed in 0.3% and 0.2% of subjects, respectively. The effects of tegaserod on the QTcF interval were not considered to be clinically meaningful.
Platelet Aggregation
There is a potential for tegaserod and its main metabolite (M29) to increase platelet aggregation in vitro. In one in vitro study, tegaserod, at concentrations up to 10 times the maximum plasma concentration (Cmax) at the recommended dose, significantly increased platelet aggregation in a concentration-dependent manner up to 74% (range 11% to 74%) compared to vehicle control (with potentiation by various agonists). In another in vitro study, M29, at concentrations up to 0.6 times the Cmax of M29 also showed a 5% to 16% increase in platelet aggregation compared to vehicle control. The clinical implications of the in vitro platelet aggregation results are unclear.
12.3. Pharmacokinetics
The pharmacokinetics of tegaserod in IBS-C patients are comparable to those in healthy subjects. The mean (±SD) peak tegaserod concentration (Cmax) was 2.9 (±1.1) ng/mL, and mean (±SD) AUC was 10.5 (±4.6) h•ng/mL following a single ZELNORM dose at 6 mg. Tegaserod systemic exposure at steady state increase proportionally over a dose range of 2 mg to 12 mg twice daily (0.3 to 2 times the approved recommended dosage). There was no significant accumulation (~10%) of tegaserod following the approved recommended dosage.
Absorption
The absolute bioavailability of tegaserod is approximately 10% when administered to fasting subjects. The median time to peak tegaserod plasma concentration (Tmax) is approximately one hour (range 0.7 to 2 hours).
Effect of Food
Compared to under fasted conditions, the tegaserod AUC was reduced by 40% to 65%, Cmax was reduced by approximately 20% to 40% and median Tmax was 0.7 hours when ZELNORM was administered 30 minutes before a high-fat, high-calorie meal (approximately 150 calories from protein, 250 calories from carbohydrates, and 500 calories from fat). Plasma concentrations were similar when ZELNORM was administered within 30 minutes prior to a meal or 2.5 hours after a meal [see Dosage and Administration (2)].
Distribution
Protein binding of tegaserod is approximately 98%. The mean volume of distribution of tegaserod (± SD) at steady-state is 368 ± 223 L following intravenous administration (ZELNORM is not approved for intravenous administration).
Elimination
The mean tegaserod terminal elimination half-life ranged from 4.6 to 8.1 hours following oral administration and the mean (± SD) plasma clearance was 77 ± 15 L/h following intravenous administration.
Metabolism
Tegaserod is metabolized via hydrolysis and direct glucuronidation. Tegaserod undergoes hydrolysis in the stomach followed by oxidation and conjugation which produces the M29 metabolite.
Excretion
Approximately two-thirds of a ZELNORM dose is excreted unchanged in the feces, with the remaining one-third excreted in the urine as metabolites.
Specific Populations
Patients with Renal Impairment
No change in the pharmacokinetics of tegaserod was observed in subjects with end stage renal disease (creatinine clearance normalized by body surface area (CrCL) <15 mL/min/1.73 m²) requiring hemodialysis. Although renal impairment does not affect the pharmacokinetics of tegaserod, the pharmacokinetics of its main metabolite (M29) are altered, the Cmax of M29 doubling and the AUC increasing 10-fold in patients with severe renal impairment (CrCL <15 mL/min/1.73 m²) compared to healthy subjects with normal renal function (CrCL >80 mL/min/1.73 m²) [see Contraindications (4), Use in Specific Populations (8.6)].
Patients with Hepatic Impairment
In subjects with mild hepatic impairment (Child-Pugh A), the mean tegaserod AUC was 31% higher and the Cmax was 16% higher compared to healthy subjects with normal hepatic function. The increase in exposure in subjects with mild impairment is not considered to be clinically relevant.
In a single subject with moderate hepatic impairment, the Cmax and AUC were 140% and 200% of that observed in healthy controls. ZELNORM has not been studied in patients with moderate or severe hepatic impairment (Child-Pugh B or C) [see Contraindications (4), Use in Specific Populations (8.7)].
Drug Interaction Studies
Effect of Other Drugs on Tegaserod
Quinidine: Coadministration of a single dose of 600 mg quinidine (P-gp inhibitor) with a single dose of ZELNORM 6 mg increased the mean tegaserod AUC(0-12h) and the mean Cmax by 50% and 44%, respectively, compared to ZELNORM administered alone. Coadministration of multiple doses of quinidine (600 mg once daily for three days) with ZELNORM 6 mg twice daily for six days increased the mean tegaserod AUC(0-12h) and Cmax by 71% and 63%, respectively, compared to ZELNORM administered alone.
Inhibitors of P-gp (e.g., ritonavir, clarithromycin, itraconazole) may modestly increase the oral bioavailability of tegaserod. The clinical relevance of increased systemic exposure as a result of P-gp inhibition is unclear.
Omeprazole: Administration of omeprazole 20 mg once daily for four days followed by ZELNORM 6 mg twice daily on day four increased the mean tegaserod AUC and Cmax by 15% and 17%, respectively, compared to ZELNORM administered alone. This increase in exposure is not considered clinically relevant.
Effect of Tegaserod on Other Drugs
No clinically significant effects of tegaserod on the pharmacokinetics of the following drugs were observed when used concomitantly with a single dose of ZELNORM 6 mg: theophylline (CYP1A2 substrate), dextromethorphan (CYP2D6 substrate), digoxin (P-gp substrate), warfarin (CYP2C9 substrate), or oral contraceptives (ethynyl estradiol and levonorgestrel).
Digoxin: Administration of a single dose of digoxin following ZELNORM 6 mg twice daily for 4 days reduced the mean Cmax and AUC of digoxin by approximately 15%. This reduction in digoxin exposure is not considered clinically relevant.
Warfarin: Coadministration of ZELNORM 6 mg twice daily with warfarin for seven days did not significantly alter the pharmacokinetics of either R- or S-warfarin or change the prothrombin time in healthy subjects.
Oral Contraceptives: Coadministration of ZELNORM 6 mg twice daily with 0.3 mg of ethinyl estradiol and 0.125 mg of levonorgestrel once daily did not affect the steady-state (Day 21) pharmacokinetics of ethinyl estradiol but reduced both the Cmax and AUC of levonorgestrel by 8%. This change in exposure is not considered clinically relevant.
In Vitro Studies Where the Drug Interaction Potential Was Not Further Evaluated Clinically
CYP enzymes:
Tegaserod does not inhibit CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2E1, and CYP3A4, and it does not induce CYP3A4 and CYP2B6.
Limited induction of CYP1A2 was observed at tegaserod concentrations in excess of 100 times the clinically relevant range.
M29 does not inhibit CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP2E1 and CYP3A4, and it does not induce CYP1A2, CYP2B6, or CYP3A4.
Transporters:
Tegaserod is a substrate for BCRP and P-gp, but not a substrate of OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3, MATE1, MATE2-K or BSEP. Drug transporter data indicated a potential inhibition of MATE1, MATE2-K, and BCRP by tegaserod at high concentrations. However, at the clinical dose of ZELNORM, a significant in vivo drug interaction via inhibition of these transporters is unlikely.
M29 is a substrate of BCRP, P-gp, OAT3 and BSEP transporters, but not a substrate of OAT1, OCT1, OCT2, OATP1B1, OATP1B3, MATE1, and MATE2-K. M29 does not inhibit the following transporters: OAT1, OAT3, OCT1, OCT2, OATP1B1, OATP1B3, MATE1, MATE2-K, BCRP, P-gp, and BSEP.
13.1. Carcinogenesis, Mutagenesis, Impairment of Fertility
Tegaserod was not carcinogenic in rats given oral dietary doses up to 180 mg/kg/day (approximately 93 to 111 times the recommended dose based on AUC) for 110 to 124 weeks.
In mice, dietary administration of tegaserod for 104 weeks produced mucosal hyperplasia and adenocarcinoma of the small intestine at 600 mg/kg/day (approximately 83 to 110 times the recommended dose based on AUC). There was no evidence of carcinogenicity at lower doses (3 to 35 times the recommended dose based on AUC).
Tegaserod was not genotoxic in the in vitro Chinese hamster lung fibroblast (CHL/V79) cell chromosomal aberration and forward mutation test, the in vitro rat hepatocyte unscheduled DNA synthesis (UDS) test or the in vivo mouse micronucleus test. The results of the Ames test for mutagenicity were equivocal.
Tegaserod at oral (dietary) doses up to 240 mg/kg/day (approximately 57 times the recommended dose based on AUC) in male rats and 150 mg/kg/day (approximately 42 times the recommended dose based on AUC) in female rats was found to have no effect on fertility and reproductive performance.
13.2. Animal Toxicology and/or Pharmacology
Inhibition of the hERG (human Ether-a-go-go-Related Gene) channel was evident only in the micromolar concentration range with an IC50 of 13 micromolar (approximately 1300 times the Cmax in humans at the recommended dose). In in vitro studies, tegaserod had no effects on impulse conduction in isolated guinea pig papillary muscle at up to 100 times the Cmax in humans, Langendorff-perfused isolated rabbit heart (QT interval) at up to 1000 times the Cmax in humans, or human atrial myocytes at multiples up to 10 times the Cmax in humans. The major metabolite, M29, had no effect on QT in the Langendorff-perfused isolated rabbit heart at multiples up to 323 times the Cmax in humans.
In anesthetized and conscious dogs, tegaserod at doses up to 92 to 134 times the recommended dose based on Cmax did not alter heart rate, QRS interval duration, QTc or other ECG parameters. In chronic toxicology studies in rats and dogs, there were no treatment-related changes in cardiac morphology after tegaserod administration at doses up to 660 times the recommended dose based on AUC.
Although tegaserod is expected to bind to 5 HT2B receptors in humans at the recommended dose, there does not appear to be any potential for heart valve injury based on functional evidence of 5 HT2B receptor antagonism.
Studies with isolated coronary and mesenteric blood vessels from non-human primates and humans showed no vasoconstrictor effect at concentrations approximately 100 times the human Cmax. Tegaserod exhibited antagonism of 5 HT-mediated vasoconstriction via 5 HT1B receptors. In rat thoracic aortic rings that were pre-constricted with phenylephrine or norepinephrine, tegaserod produced vasorelaxation, with IC50 values 6 and 64 times the Cmax plasma concentrations in humans, respectively. No effects were observed in the basal tone of aortic rings at concentrations up to 1000 times the human Cmax.
In studies with an anesthetized rat model for measuring macro- and micro-circulation of the colon, intraduodenal dosing with tegaserod (approximately 7 times the recommended dose based on Cmax) produced no clinically relevant effect on blood pressure, heart rate, or vascular conductance.
14. Clinical Studies
Results in Women
ZELNORM is not recommended in females 65 years of age and older with IBS-C [see Indications and Dosage (1)].
In three multicenter, double-blind, placebo-controlled trials, 2,470 women (mean age 43 years [range 17 to 89 years]; 86% Caucasian, 10% African American) with at least a 3-month history of IBS-C symptoms prior to the baseline period that included abdominal pain, bloating and constipation received either ZELNORM 6 mg twice daily or placebo. In all patients, constipation was characterized by at least two of the following three symptoms each occurring >25% of the time over a 3-month period: <3 bowel movements/week, hard or lumpy stools, or straining with a bowel movement.
The design for the three trials consisted of a 4-week placebo-free baseline period followed by a 12-week double-blind treatment period. Studies 1 and 2 evaluated a fixed dose regimen of tegaserod 6 mg twice daily while Study 3 utilized a dose-titration design.
Each week of the 4-week placebo-free baseline period and the 12-week double-blind treatment period, patients were asked the question, "Please consider how you felt this past week in regard to your IBS, in particular your overall well-being, and symptoms of abdominal discomfort, pain and altered bowel habit. Compared to the way you usually felt before entering the trial, how would you rate your relief of symptoms during the past week?" The response variable consisted of the following five categories: completely relieved, considerably relieved, somewhat relieved, unchanged, or worse. Patients were classified as responders within a month if they were considerably or completely relieved for at least two of the four weeks, or if they were at least somewhat relieved for each of the four weeks.
Calculated response rates during month 1 and during month 3, as described above, are shown in Table 3. The differences in response rates vs. placebo were greater at month 1 than month 3.
Table 3. Efficacy Responder* Rates in the Three Placebo-Controlled IBS-C Trials:
| Study | Month 1 | Month 3** | ||||
|---|---|---|---|---|---|---|
| Proportion of Responders (Female) | Proportion of Responders (Female) | |||||
| ZELNORM 6 mg twice daily | Placebo | Difference [95% CI] | ZELNORM 6 mg twice daily | Placebo | Difference [95% CI] | |
| 1 | 76/244 | 42/240 | 14% | 95/244 | 66/240 | 11% |
| (31%) | (17%) | [6%,21%] | (39%) | (28%) | [3%,20%] | |
| 2 | 265/767 | 164/752 | 13% | 334/767 | 292/752 | 5% |
| (35%) | (22%) | [8%,17%] | (44%) | (39%) | [0%,10%] | |
| 3 | 80/233 | 47/234 | 14% | 100/233 | 88/234 | 5% |
| (34%) | (20%) | [6%,22%] | (43%) | (38%) | [-4%,14%] | |
* A responder is defined as a patient with ≥2 of 4 weeks with complete or considerable relief or 4 of 4 weeks with at least somewhat relief during the last 4 available weeks.
** Primary efficacy assessment.
In a subgroup of female patients less than 65 years of age (90%, 97%, and 91% of all female patients in Studies 1, 2 and 3, respectively), the treatment differences were generally similar at both month 1 and month 3 to the overall results shown in Table 3.
The same efficacy variable (i.e., complete relief, considerable relief, somewhat relief, unchanged, worse) was analyzed on a weekly basis. The proportion of all female patients with complete, considerable or somewhat relief at weeks 1, 4, 6, 8 and 12 are shown in Figure 1 below.
Figure 1. Weekly Proportion of Patients with Somewhat, Considerably and Complete Relief in the Three Placebo-Controlled IBS-C Trials:
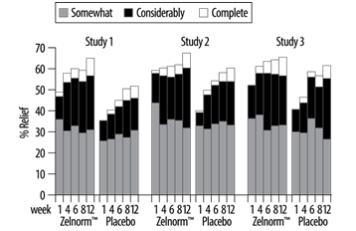
In addition, individual symptoms of abdominal pain/discomfort and bloating were assessed daily using a six or seven point intensity scale. A positive response was defined as at least a 1-point reduction in the scale. During the first four weeks in the fixed dose trials, 8% to 11% more ZELNORM-treated patients than placebo-treated patients were responders for abdominal pain/discomfort. Similarly, 9% to 12% more ZELNORM-treated patients were responders for bloating. Corresponding differences at month 3 were 1% to 10% responders for abdominal pain/discomfort and 4% to 11% responders for bloating. Patients on ZELNORM also experienced an increase in median number of bowel movements from 3.8/week at baseline to 6.3/week at month 1 and 6.0/week at month 3, while placebo patients increased from 4.0/week to 5.1/week at month 1 and 5.5/week at month 3.
Results in Men
In two randomized, placebo-controlled, double-blind trials enrolling 288 males, efficacy response rates were similar between ZELNORM and placebo in the male subgroup [see Indications and Usage (1)].
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.