Source: European Medicines Agency (EU) Revision Year: 2018 Publisher: BIOGEN IDEC Limited, Innovation House, 70 Norden Road, Maidenhead, Berkshire, SL6 4AY, United Kingdom
Pharmacotherapeutic group: immunosuppressants, interleukin inhibitors
ATC code: L04AC01
Daclizumab beta is a humanised IgG1 monoclonal antibody that binds to CD25 (IL-2Rα), and prevents IL-2 binding to CD25. Daclizumab beta modulates IL-2 signalling by blocking CD25-dependent, high-affinity IL-2 receptor signalling, resulting in higher levels of IL-2 available for signalling through the intermediate-affinity IL-2 receptor. Key effects of this IL-2 pathway modulation potentially related to the therapeutic effects of daclizumab beta in MS include selective antagonism of activated T-cell responses, and expansion of immunoregulatory CD56bright natural killer (NK) cells, which have been shown to selectively decrease activated T-cells. Together, these immunomodulatory effects of daclizumab beta are believed to reduce CNS pathology in MS and thereby reduce the occurrence of relapses and disability progression.
In clinical studies, the pharmacodynamic effects of Zinbryta 150 mg administered subcutaneously every 4 weeks were consistent with modulation of IL-2 signalling as evidenced by the rapid and sustained saturation of the target CD25 receptors on circulating T-cells and a sustained approximately 2-fold increase in serum IL-2 concentration. In addition, an increase in CD56bright NK cells and a decrease in regulatory T-cells (defined as CD4+ CD127lowFoxP3+ T-cells) was observed within 2 weeks after the first dose, with a sustained 5-fold increase in CD56bright NK cells above baseline and an approximately 60% decrease in regulatory T-cells in the treatment phase, with a return to baseline levels approximately 20-24 weeks after the last dose. During Zinbryta treatment, mean cell counts for the major immune subsets (T, B, and NK cells) remained within normal ranges; total lymphocyte, T and B cell counts decreased on average ≤10% from baseline during the first year of treatment. Total lymphocyte counts returned to baseline levels approximately 8-12 weeks after the last dose of Zinbryta (150mg). Total lymphocyte counts <0.8x109 cells/L ([Common Terminology Criteria for Adverse Events – CTCAE] Grade 2; at least one measurement) occurred in 4% of placebo-treated and 5% of Zinbryta-treated patients in the SELECT study, and 9% of the interferon beta-1a (intramuscular)-treated and 8% of Zinbryta-treated patients in the DECIDE study. Total NK cell counts increased approximately 1.5-fold as a result of the change in CD56bright NK cells.
The efficacy of Zinbryta was demonstrated in two studies (SELECT and DECIDE) in patients with RMS. The SELECT study was double-blind, randomised, placebo-controlled, with either Zinbryta 150 mg (n=208), or 300 mg (n=209) versus placebo (n=204) every 4 weeks for 52 weeks. The DECIDE study was double-blind, randomised, parallel-group, active-controlled with Zinbryta 150 mg every 4 weeks (n=919) versus interferon beta-1a (intramuscular) 30 micrograms weekly (n=922), for a minimum of 2 to a maximum of 3 years (96 to 144 weeks). The study designs and baseline characteristics are presented in Table 3.
Table 3. Study design and baseline characteristics for SELECT study and DECIDE study:
| Study name | SELECT | DECIDE |
|---|---|---|
| Study design | ||
| Treatment | 52 weeks | 96 to 144 weeks |
| Disease history | Patients with RMS, at least 1 relapse (clinical and/or MRI) during the year prior to randomisation, and had an EDSS score between 0 to 5.0. For DECIDE, at least 2 relapses (one of which was a clinical relapse) within the prior 3 years was also required. | |
| Baseline characteristics | ||
| Mean age (years) | 35.7 | 36.3 |
| Mean disease duration (years) | 4.1 | 4.2 |
| Mean number of relapses within 12 months prior to study | 1.4 | 1.6 |
| Median EDSS score | 2.5 | 2.0 |
| Percent with EDSS ≥3.5 | 36% | 30% |
| Percent with ≥1 Gd enhancing lesion (mean) | 44% (1.8) | 46% (2.1) |
| Percent ≥2 relapses in the year prior to study | 31% | 46% |
| Percent prior DMT use (%) | 20% | 41% |
Results for the SELECT study are shown in Table 4. Treatment with Zinbryta 150 mg every 4 weeks versus placebo significantly reduced the annualised relapse rate (ARR) and risk of relapse compared to placebo. In addition, there was a statistically significant effect on 24 week confirmed disability progression in Zinbryta treated patients with a hazard ratio 0.24 [95% CI: 0.09, 0.63]. The 300 mg dose did not provide additional benefit over the 150 mg dose.
Table 4. SELECT study clinical and MRI results (at 52 weeks):
| Placebo | Zinbryta 150 mg | p-value | |
|---|---|---|---|
| Clinical endpoints | |||
| Number of patients | 196 | 201 | |
| Annualised relapse rate | 0.458 | 0.211 | |
| Rate ratio [95% CI] | 0.461 [0.318, 0.668] | p<0.0001 | |
| Percentage of patients relapse-free | 64% | 81% | |
| Hazard ratio* [95% CI] | 0.45 [0.30, 0.67] | p<0.0001 | |
| Percentage with 24 weeks confirmed disability progression | 11% | 2.6% | |
| Hazard ratio [95% CI] | 0.24 [0.09, 0.63] | p=0.0037 | |
| Percentage with 12 weeks confirmed disability progression | 13% | 6% | |
| Hazard ratio [95% CI] | 0.43 [0.21, 0.88] | p=0.0211 | |
| Mean change in MSIS-29 physical score | 3.0 point worsening | 1.0 point improvement | p=0.0008 |
| MRI endpoints# | |||
| Mean number of new or newly enlarging T2 hyperintense lesions | 8.13 | 2.4 | |
| Lesion mean ratio [95% CI] | 0.30 [0.22, 0.40] | p<0.0001 | |
| Mean number of new T1 Gdenhancing lesions between 8 and 24 weeks (on monthly MRI scans) | 4.79 | 1.46 | |
| Lesion mean ratio [95% CI] | 0.31 [0.20, 0.48] | p<0.0001 | |
* Hazard ratio for the risk of relapse
# MRI analyses used evaluable dataset for each endpoint; T1 Gd-enhancing: MRI intensive population
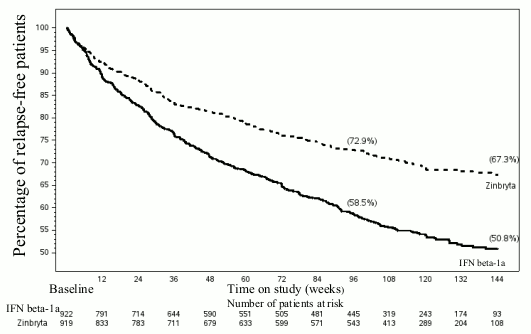
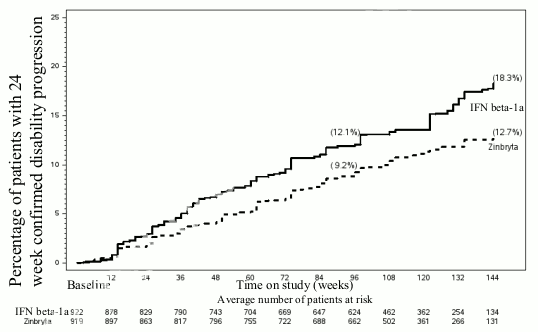
Table 5 and Figures 1-2 show the results for the DECIDE study. Zinbryta significantly reduced the ARR and the risk of relapse, compared to interferon beta-1a (intramuscular)-treated patients. In addition, there was a statistically significant effect on 24 week confirmed disability progression in Zinbryta treated patients with a hazard ratio 0.73 [95% CI: 0.55, 0.98]. At week 96, Zinbryta demonstrated a statistically significant reduction in the number of new or newly enlarging T2 hyperintense lesions, the number of new T1 Gd-enhancing lesions and the mean number of new T1 hypointense lesions. In addition, Zinbryta reduced clinically meaningful worsening in the patientreported physical impact of MS (≥7.5 point worsening from baseline to week 96 in the MSIS-29 physical score) compared to interferon beta-1a (intramuscular).
Table 5. DECIDE study clinical and MRI results (96 to 144 weeks) (Values refer to results at 96 weeks, unless otherwise indicated):
| Interferon beta-1a (intramuscular) 30 micrograms | Zinbryta 150 mg | p-value | |
|---|---|---|---|
| Clinical endpoints | |||
| Number of patients | 922 | 919 | |
| Annualised relapse rate* | 0.393 | 0.216 | |
| Rate ratio* [95% CI] | 0,550 [0.469, 0.645] | p<0.0001 | |
| Percentage of patients relapse-free | 59% | 73% | |
| Hazard ratio#* [95% CI] | 0.59 [0.50, 0.69] | p<0.0001 | |
| Percentage with 24 weeks confirmed disability progression | 12% | 9% | |
| Hazard ratio* [95% CI] | 0.73 [0.55, 0.98] | p=0.03 | |
| Percentage with 12 weeks confirmed disability progression | 14% | 12% | |
| Hazard ratio* [95% CI] | 0.84 [0.66, 1.07] | p=0.16 | |
| Percentage of patients with clinically meaningful worsening in MSIS-29 physical score (≥7.5 point) | 23% | 19% | |
| Odds ratio [95% CI] | 0.76 [0.60, 0.95] | p=0.018 | |
| MRI endpoints† | |||
| Mean number of new or newly enlarging T2 hyperintense lesions | 9.44 | 4.31 | |
| Lesion mean ratio [95% CI] | 0.46 [0.39, 0.53] | p<0.0001 | |
| Mean number of new T1 Gd-enhancing lesions | 1.0 | 0.4 | |
| Odds ratio [95% CI] | 0.25 [0.20, 0.32] | p<0.0001 | |
| Mean number of new T1 hypointense lesions | 4.43 | 2.13 | |
| Lesion mean ratio [95% CI] | 0.48 [0.42, 0.55] | p<0.0001 | |
* Rates and risk reductions/endpoints are calculated over the treatment period up to 144 weeks.
# Hazard ratio for the risk of relapse.
† MRI analyses used evaluable dataset for each MRI endpoint.
Subgroup analyses of the SELECT and DECIDE studies demonstrated a consistent effect of Zinbryta compared to placebo and interferon beta-1a (intramuscular) across subgroups defined by demographic and MS disease characteristics. In the DECIDE study subgroup analysis, there was a statistically significant reduction observed compared to interferon beta-1a (intramuscular) on ARR and the number of new or newly enlarging T2 hyperintense lesions across subgroups (gender, age, prior MS DMT therapy, and disease activity levels).
Although the effect on disability progression was mainly seen in patients with baseline EDSS <3.5, evidence of efficacy was shown in patients with relapsing secondary progressive MS (SPMS) as defined by baseline EDSS ≥3.5 and at least one of the three: confirmed 24 week worsening of EDSS, or ≥20% decline on Timed 25-foot Walk (T25FW), or, ≥20% decline on 9-Hole Peg Test (9-HPT).
Highly active disease was defined as follows:
Clinical trial data from the DECIDE study demonstrated consistent treatment effects in the highly active disease subgroup. Compared with interferon beta-1a intramuscular (n=440), Zinbryta (n=404) led to reductions on ARR (rate ratio 0.52 [95% CI: 0.42, 0.64], p<0.0001), number of new or newly enlarging T2 hyperintense lesions (lesion mean ratio 0.46 [95% CI: 0.37, 0.57], p<0.0001), and 24 weeks confirmed disability progression (hazard ratio 0.60 [95% CI: 0.40, 0.89], p=0.012).
Figure 1. Percentage of patients relapse-free (DECIDE study):
Figure 2. Proportion of patients with 24 week confirmed disability (DECIDE study):
The European Medicines Agency has deferred the obligation to submit the results of studies with Zinbryta in one or more subsets of the paediatric population in treatment of multiple sclerosis (see section 4.2 for information on paediatric use).
Daclizumab beta pharmacokinetics are well described by a two-compartment model with first-order absorption and elimination.
Following subcutaneous administration of daclizumab beta, the median time to reach maximum serum concentrations (Tmax) ranged from 5 to 7 days. The absolute bioavailability of daclizumab beta 150 mg subcutaneously administered was approximately 90% based on a cross-study population pharmacokinetic analysis of subcutaneous and intravenous dosing.
Following subcutaneous administration of daclizumab beta 150 mg every 4 weeks, steady-state serum daclizumab beta concentrations were achieved by the 4th dose and daclizumab beta accumulated to a level approximately 2.5-fold compared to a single dose. At steady state, daclizumab beta mean maximum serum concentration (Cmax), minimum serum concentration (Cmin) and area under the serum concentration-time curve over the dosing interval (AUCtau) values were approximately 30 micrograms/mL, 15 micrograms/mL and 640 day*micrograms/mL, respectively, with inter-patient variability (% CV) of approximately 40%.
Based on the cross-study population pharmacokinetic analysis, the steady-state volume of distribution of daclizumab beta is 6.34 L in a patient with a body weight of 68 kg (approximate median of evaluated patients). This small volume of distribution indicates that daclizumab beta is primarily confined to the vascular and interstitial spaces.
The exact metabolic pathway for daclizumab beta has not been characterised. As an IgG1 monoclonal antibody, daclizumab beta is expected to undergo catabolism to peptides and amino acids in the same manner as endogenous IgG. Daclizumab beta is not expected to undergo metabolism by hepatic enzymes such as CYP isoenzymes (see section 4.5).
As an IgG1 monoclonal antibody, daclizumab beta is not expected to undergo renal elimination.
Based on the cross-study population pharmacokinetic analysis, the clearance of daclizumab beta is 0.212 L/day with a terminal half-life value of approximately 21 days. Daclizumab beta clearance in patients who developed neutralising antibodies was, on average, 19% higher (see section 4.8 Immunogenicity).
Consistent with results from individual studies, a cross-study population pharmacokinetic analysis indicated that daclizumab beta exposure is more than dose-proportional in the 50 mg to 100 mg subcutaneous dose range and is dose proportional in the 100 mg to 300 mg subcutaneous dose range.
Within the studied regimens of daclizumab beta 150 mg and 300 mg administered subcutaneous every 4 weeks in MS patients, there was no clear relationship between daclizumab beta exposure and clinical efficacy endpoints (ARR, T2 lesions and Gd-enhancing lesions) or safety endpoints of interest (serious infection status, moderate or severe cutaneous adverse reaction, and AST/ALT >5 times the ULN).
No studies were conducted to evaluate daclizumab beta pharmacokinetics in patients with renal or hepatic impairment. Daclizumab beta is not expected to undergo renal elimination or metabolism by hepatic enzymes (see section 4.2).
Based on the cross-study population pharmacokinetic analysis, body weight accounted for less than 40% of the inter-patient variability in daclizumab beta clearance. No meaningful differences in clinical efficacy or safety were observed among the subgroups of MS patients by weight quartile in the DECIDE study.
Based on the cross-study population pharmacokinetic analysis, daclizumab beta pharmacokinetics were not influenced by age (range: 18 to 66 years; n=1670) or gender (n = 567 males and 1103 females).
No pharmacokinetic differences were observed between Japanese and Caucasian healthy volunteers.
Preclinical safety studies were conducted in cynomolgus monkeys due to species specificity of daclizumab beta binding only to human or primate CD25.
Carcinogenicity studies with daclizumab beta have not been conducted. In two 9 month studies in monkeys there were no pre-neoplastic or neoplastic tissue observed.
Genotoxicity studies have not been conducted.
Daclizumab beta did not affect reproductive capacity in female and male cynomolgus monkeys (AUC in females and males up to 85 and 100 times higher than the exposure at the clinical dose respectively). There was no effect on foetal development and no evidence of teratogenicity. Daclizumab beta had no effect on peri- and post-natal development from birth to up to 6 months in the offspring. Exposures (AUC) in these studies ranged from 55 to 140 times that observed with the clinical dose. Daclizumab beta was detected in the milk of 11/14 lactating monkeys at levels that were <0.122% of the maternal serum levels, with no adverse reactions observed in the off-spring.
In two 9 month studies conducted in cynomolgus monkeys daclizumab beta was subcutaneously administered at bi-weekly doses of 10-200 mg/kg. Chronic administration of daclizumab beta at all doses increased the incidence of skin findings (compared to those observed in control animals). These findings (dry, red raised patchy areas of the skin, as compared to controls, that correlated microscopically with acanthosis/hyperkeratosis and subacute to chronic inflammation) were characterised predominantly as mild to moderate, with one case assessed as severe.
A dose dependent increase in incidence of microglial aggregates above background was observed in the brain and spinal cord of monkeys treated with ≥35 mg/kg, (AUC 27 times higher than the clinical dose). Following a recovery period of up to 12 weeks, there was evidence of reversibility. Microglial aggregates in monkeys did not increase in incidence or severity with increased duration of dosing and were not associated with neuronal damage or neurobehavioral effects. A small subset of microglial aggregates were associated with microhaemorrhage but with no evident functional sequelae in monkeys.
In vitro investigative studies suggest that microglial aggregates are not due to a direct effect of daclizumab beta on microglial cells but are likely to be attributable to an increase in local IL-2 bioavailability.
The clinical relevance of microglial aggregates is unknown, however no deleterious neurologic effects attributed to the microscopic change have been observed in monkeys.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.