ZURAMPIC Film-coated tablet Ref.[7621] Active ingredients: Lesinurad
Source: European Medicines Agency (EU) Revision Year: 2017 Publisher: Grünenthal GmbH, Zieglerstr. 6, 52078, Aachen, Germany, Tel.: +49-241-569-0
Pharmacodynamic properties
Pharmacotherapeutic group: Antigout preparations, preparations increasing uric acid excretion
ATC code: M04AB05
Mechanism of action
Lesinurad is a selective uric acid reabsorption inhibitor that inhibits uric acid transporter URAT1. URAT1 is responsible for the majority of the reabsorption of filtered uric acid from the renal tubular lumen. By inhibiting URAT1, lesinurad increases uric acid excretion and thereby lowers serum uric acid (sUA). Lesinurad also inhibits OAT4, a uric acid transporter involved in diuretic-induced hyperuricaemia.
Lesinurad, when combined with a xanthine oxidase inhibitor, increases uric acid excretion and decreases uric acid production resulting in greater sUA lowering. Lesinurad should only be used in combination with a xanthine oxidase inhibitor because combination use reduces the amount of uric acid available for excretion and decreases the risk of renal-related events.
Pharmacodynamic effects
Effects on serum uric acid and urinary excretion of uric acid
In healthy subjects, lesinurad 200 mg lowered sUA levels and increased renal clearance and fractional excretion of uric acid. Mean sUA reductions following Zurampic 200 mg administration alone were approximately 46% and 26% at 6 hours and 24 hours post-dose, respectively. When Zurampic 200 mg was added to a xanthine oxidase inhibitor (i.e. febuxostat), additional 25% and 19% of sUA reductions were observed at 6 hours and 24 hours post-dose, respectively.
Effect on cardiac repolarisation
Lesinurad at doses up to 1,600 mg did not demonstrate an effect on ECG parameters (including QTc interval) in healthy subjects.
Clinical efficacy and safety
The efficacy of Zurampic 200 mg and 400 mg once daily was studied in 3 multicentre, randomised, double-blind, placebo-controlled clinical studies in 1,537 adult patients (13% of these patients were elderly, ≥65 years old) with hyperuricaemia and gout in combination with a xanthine oxidase inhibitor, allopurinol (CLEAR1 and CLEAR2) or febuxostat (CRYSTAL). All studies were of 12 months duration and patients received prophylaxis for gout flares with colchicine or NSAIDs during the first 5 months of lesinurad treatment.
Based on these studies, Zurampic is only recommended at a dose of 200 mg once daily in combination with a xanthine oxidase inhibitor (see sections 4.2 and 4.4).
Zurampic as add-on to allopurinol in inadequate responders
CLEAR1 and CLEAR2 enrolled patients with gout who were on a stable dose of allopurinol of at least 300 mg (or 200 mg for moderate renal impairment), had serum uric acid levels greater than 6.5 mg/dL and reported at least 2 gout flares in the previous 12 months. Across both studies, 61% of patients had mild or moderate renal impairment and 19% had tophi at baseline. Patients continued their allopurinol dose and were randomised 1:1:1 to receive Zurampic 200 mg, Zurampic 400 mg, or placebo once daily.
The primary efficacy endpoint in both CLEAR1 and CLEAR2 was the proportion of patients achieving a serum uric acid target level of less than 6 mg/dL by Month 6. In both studies, significantly more patients treated with Zurampic 200 mg in combination with allopurinol achieved the target serum uric acid level of less than 6 mg/dL by Month 6 and by Month 12 compared with patients receiving placebo in combination with allopurinol (see Table 2).
The stability of the sustained response was demonstrated with a greater proportion of patients treated with Zurampic 200 mg in combination with allopurinol achieving the target serum uric acid level at each visit for 3 consecutive months (Months 4, 5 and 6) compared to patients treated with placebo in combination with allopurinol (see Table 2).
Table 2. Proportion of patients who achieved target serum uric acid levels (<6 mg/dL) with Zurampic in combination with allopurinol - Pooled data from CLEAR1 and CLEAR2 studies:
| Proportion of patients who met serum uric acid target (<6.0 mg/dL) N (%) | Difference in proportion (95% C.I.) | ||
|---|---|---|---|
| Timepoint | Placebo + allopurinol N=407 | Zurampic 200 mg + allopurinol N=405 | Zurampic 200 mg vs. placebo |
| Months 4, 5, 6 | 48 (12%) | 155 (38%) | 0.26 (0.21, 0.32) |
| Month 6 | 104 (26%) | 222 (55%) | 0.29 (0.23, 036) |
| Month 12 | 105 (26%) | 203 (50%) | 0.24 (0.18, 0.31) |
Zurampic when added to allopurinol caused an immediate reduction of the mean serum uric acid levels, as compared to placebo, which was sustained in the long term in those patients who continued treatment (see Figure 1).
Figure 1. Mean serum uric acid levels in pooled clinical studies with Zurampic in combination with allopurinol in patients with inadequate response (sUA ≥6 mg/dL) to allopurinol alone:
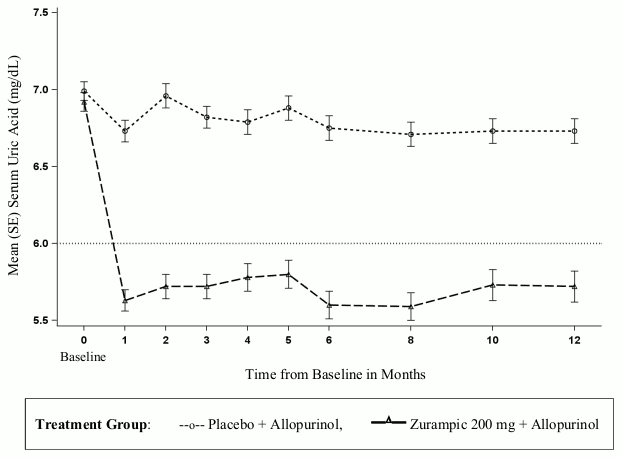
In each of the studies, a greater proportion of patients treated with Zurampic 200 mg in combination with allopurinol compared with placebo in combination with allopurinol achieved a serum uric acid less than 5 mg/dL by Month 6 (CLEAR1: 29% versus 10%; CLEAR2: 35% versus 5%).
Zurampic in combination with febuxostat in tophaceous gout
CRYSTAL enrolled gout patients with measurable tophi. Patients received febuxostat 80 mg once daily for 3 weeks and then were randomised 1:1:1 to once daily doses of Zurampic 200 mg, Zurampic 400 mg, or placebo in combination with febuxostat. Sixty-six percent of patients had mild or moderate renal impairment. Fifty percent of patients had a baseline sUA ≥5.0 mg/dL, which was after 3 weeks of treatment with febuxostat alone.
Zurampic when added to febuxostat caused an immediate reduction of the mean serum uric acid levels, as compared to placebo, which was sustained in the long term in those patients who continued treatment.
In the subgroup of patients with a baseline sUA ≥5.0 mg/dL, after 3 weeks of febuxostat therapy, a significant difference was achieved at all study visits for Zurampic 200 mg in combination with febuxostat compared with placebo in combination with febuxostat (see Table 3).
Table 3. Proportion of patients with baseline sUA ≥5.0 mg/dLwho achieve target serum uric acid levels (<5 mg/dL) with Zurampic in combination with febuxostat:
| Proportion of patients who met serum uric acid Target (<5.0 mg/dL) N (%) | Difference in proportion (95% C.I.) | ||
|---|---|---|---|
| Timepoint | Placebo + febuxostat 80 mg N=51 | Zurampic 200 mg + febuxostat 80 mg N=59 | Zurampic 200 mg vs. placebo |
| Months 4, 5, 6 | 6 (12%) | 23 (39%) | 0.27 (0.12, 0.42) |
| Month 6 | 12 (24%) | 26 (44%) | 0.21 (0.03, 0.38) |
| Month 12 | 12 (24%) | 27 (46%) | 0.22 (0.05, 0.39) |
Primary end-point in patients with renal impairment
Consistent with the overall population, the proportion of patients with mild to moderate renal impairment (eCrCL 30-89 mL/min) who achieved target serum uric acid levels at Month 6 was 56% for Zurampic 200 mg versus 29% for placebo when added to allopurinol, and 40% for Zurampic 200 mg versus 26% for placebo when added to febuxostat in patients with baseline sUA ≥5.0 mg/dL.
Clinical outcomes - gout flares requiring treatment
The rates of gout flare requiring treatment were low and comparable to placebo in the last 6 months of the randomised trials (after gout flare prophylaxis was discontinued) with median scores of zero. In the long-term uncontrolled extension trials, the rates of gout flares requiring treatment further decreased in the 60% of subjects who entered the extension studies and continued treatment with Zurampic 200 mg in combination with allopurinol or febuxostat for up to an additional year of treatment.
Clinical outcomes - tophus resolution and reduction
In CRYSTAL, the proportion of subjects who experienced a complete resolution (defined as 100% resolution of at least one target tophus and no single tophus showing progression) of ≥1 target tophus was higher in the group treated with Zurampic 200 mg in combination with febuxostat compared with placebo in combination with febuxostat, although the difference was not statistically different (26% compared with 21%). After continued treatment of up to 24 months on Zurampic 200 mg in combination with febuxostat, the proportion of subjects who experienced complete resolution of at least one target tophus increased to 53% of subjects.
Paediatric population
The European Medicines Agency has waived the obligation to submit the results of studies with Zurampic in all subsets of the paediatric population for the treatment and prevention of hyperuricaemia (see section 4.2 for information on paediatric use).
Pharmacokinetic properties
Absorption
The absolute bioavailability of lesinurad is approximately 100%. Lesinurad is rapidly absorbed after oral administration. Following administration of a single oral dose of lesinurad in either the fed or fasted state, maximum plasma concentrations (Cmax) were attained within 1 to 4 hours. Cmax and AUC exposures of lesinurad increased proportionally with single doses of lesinurad from 5 to 1,200 mg. In the fed state, after a single dose of lesinurad 200 mg, geometric mean lesinurad Cmax and AUC were 6 μg/mL and 29 μg/hr/mL, respectively. There was no apparent influence of the fat content in the meal on the pharmacokinetics of lesinurad. In clinical trials, Zurampic was administered with food, because the serum uric acid lowering was improved under fed conditions.
Zurampic is administered as a 50:50 mixture of lesinurad atropisomers. The ratio of atropisomer 1 to atropisomer 2 AUC(0-24) was 44:56 because atropisomer 1 undergoes more extensive metabolism than atropisomer 2, causing atropisomer 1 to have lower plasma exposure than atropisomer 2.
Distribution
Lesinurad is extensively bound to proteins in plasma (greater than 98%), mainly to albumin. Plasma protein binding is not meaningfully altered in patients with renal or hepatic impairment. The mean steady state volume of distribution of lesinurad was approximately 20 L following intravenous dosing. Mean plasma-to-blood ratios of lesinurad AUC and Cmax were approximately 1.8, indicating that radioactivity was largely contained in the plasma space and did not penetrate or partition extensively into red blood cells.
Biotransformation
Lesinurad undergoes oxidative metabolism mainly via cytochrome P450 (CYP) 2C9 to intermediate metabolite M3c (not detected in vivo) and is subsequently metabolised by mEH to metabolite M4; there is minimal contribution from CYP1A1, CYP2C19, and CYP3A to the metabolism of lesinurad. Atropisomer 1 is extensively metabolised by CYP2C9 whereas atropisomer 2 is minimally metabolised by both CYP2C9 and CYP3A4. It is unclear if metabolite plasma exposures are minimal. Metabolites are not known to contribute to the uric acid lowering effects of lesinurad.
Elimination
Renal clearance is 25.6 mL/min (CV=56%). Lesinurad is highly protein bound and renal clearance is high (as compared to typical human glomerular filtration rate), indicating that active secretion plays an important role in the renal excretion of lesinurad. Within 7 days following single dosing of radiolabelled lesinurad, 63% of administered radioactive dose was recovered in urine and 32% of administered radioactive dose was recovered in faeces. Most of the radioactivity recovered in urine (>60% of dose) occurred in the first 24 hours. Unchanged lesinurad in urine accounted for approximately 30% of the dose. The elimination half-life (t½) of lesinurad was approximately 5 hours following a single dose. Lesinurad does not accumulate following multiple doses.
Linearity/non-linearity
Following multiple once daily dosing of Zurampic, there was no evidence of time dependent changes in pharmacokinetic properties and dose proportionality was preserved.
In vitro assessment of interactions
Lesinurad is mainly metabolised by CYP2C9 and mEH, and to a lesser extent by CYP1A1, CYP2C19 and CYP3A. In vitro, lesinurad is an inhibitor of CYP2C8, but not of CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6, CYP3A4 and mEH. In addition, lesinurad is an in vitro inducer of CYP2B6 and CYP3A via CAR/PXR. In vivo, lesinurad is neither an inhibitor nor an inducer of CYP2C9 and 2C8, but a mild to moderate inducer of CYP3A. CYP2B6 has not been studied in vivo.
Lesinurad is a substrate of OATP1B1, OAT1, OAT3 and OCT1. In vitro, lesinurad is an inhibitor of OATP1B1, OAT1, OAT3, OAT4 and OCT1 at clinically relevant plasma concentrations. However, the in vivo activity of OATP1B1, OAT1, OAT3 and OCT1 was not affected by lesinurad. Lesinurad is not an in vitro inhibitor of P-glycoprotein, BCRP, OATP1B3, MRP2, MRP4, OCT2, MATE1 and MATE2-K.
Special populations
Renal impairment
The population pharmacokinetic analysis of clinical data in gout patients treated for up to 12 months estimated increases in lesinurad exposure of approximately 12%, 31% and 65% in patients with mild, moderate, and severe renal impairment, respectively, compared with patients with normal renal function.
Following administration of a single dose of lesinurad to individuals with renal impairment compared to those with normal renal function lesinurad Cmax and AUC, respectively, were 36% and 30% higher (200 mg) in patients with mild renal impairment (eCrCL 60 to 89 mL/min), 20% and 73% higher (200 mg) and 3% and 50% higher (400 mg) in patients with moderate renal impairment (eCrCL 30 to 59 mL/min), and 13% higher and 113% higher (400 mg) in patients with severe renal impairment (eCrCL <30 mL/min).
Hepatic impairment
Following administration of a single dose of lesinurad at 400 mg in patients with mild (Child-Pugh class A) or moderate (Child-Pugh class B) hepatic impairment, lesinurad Cmax was comparable and lesinurad AUC was 7% and 33% higher, respectively, compared to individuals with normal hepatic function. There is no clinical experience in patients with severe (Child-Pugh class C) hepatic impairment.
CYP2C9 poor metabolisers
Approximately half of an oral dose of lesinurad is cleared via CYP2C9 metabolism. The effect of CYP2C9 genotype on the pharmacokinetics of lesinurad was studied in 8 healthy subjects and 59 patients with gout following daily dosing of lesinurad ranging from 200 mg to 600 mg in the absence or presence of a xanthine oxidase inhibitor. At the 400 mg dose, when compared with extensive CYP2C9 metabolisers (CYP2C9 *1/*1 [N=41]), increased lesinurad exposures were observed in intermediate CYP2C9 metabolisers (CYP2C9 *1/*3 [N=4], approximately 22% increase in AUC) and in poor CYP2C9 metabolisers (CYP2C9 *3/*3 [N=1], approximately 111% increase in AUC) accompanied with higher lesinurad renal excretion. However, individual values were well within the range observed in the extensive metaboliser subjects.
Patients who are known or suspected to be CYP2C9 poor metabolisers based on previous history or experience with other CYP2C9 substrates should use Zurampic with caution (see section 4.4).
Other special populations
Based on population pharmacokinetic analysis, age, gender, race and ethnicity do not have a clinically meaningful effect on the pharmacokinetics of lesinurad. Based on pharmacokinetic modelling simulations, patients with moderate renal impairment and reduced CYP2C9 activity (co-administration of a CYP2C9 inhibitor or a CYP2C9 poor metabolizer) are predicted to have an increase in AUC of approximately 200% in comparison to normal renal function and unimpaired CYP2C9 activity.
Preclinical safety data
Non-clinical data reveal no special hazard for humans based on conventional studies of safety pharmacology, repeated dose toxicity, genotoxicity, carcinogenic potential, toxicity to reproduction and development.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.