Bexagliflozin
Chemical formula: C₂₄H₂₉ClO₇ Molecular mass: 464.16 g/mol PubChem compound: 25195624
Mechanism of action
Bexagliflozin is an inhibitor of sodium-glucose co-transporter 2 (SGLT2), the transporter responsible for reabsorption of the majority of glucose from the renal glomerular filtrate in the renal proximal tubule. By inhibiting SGLT2, bexagliflozin reduces renal reabsorption of filtered glucose and lowers the renal threshold for glucose, and thereby increases urinary glucose excretion.
Pharmacodynamic properties
Urinary Glucose Excretion and Urinary Volume
Dose-dependent increases in urinary glucose excretion (UGE) accompanied by increases in urine volume were observed in healthy subjects and in adults with type 2 diabetes mellitus following single- and multiple-dose administration of bexagliflozin. Dose-response analysis indicates that 20 mg bexagliflozin provides near-maximal UGE. Elevated UGE was maintained after multiple-dose administration.
Cardiac Electrophysiology
At 5 times the recommended dose, bexagliflozin does not prolong the QTc interval to any clinically significant extent.
Pharmacokinetic properties
The pharmacokinetics of bexagliflozin are similar in healthy subjects and adults with type 2 diabetes mellitus. Following dosing in the fasted state, mean Cmax and AUC0-∞ were 134 ng/mL and 1,162 ng·h/mL, respectively. Bexagliflozin does not exhibit time-dependent pharmacokinetics and accumulates in plasma up to ~20% following multiple dosing.
Absorption
Following oral administration of bexagliflozin, peak plasma concentrations of bexagliflozin were reached between 2–4 hours post-dose and can be delayed if taken after a meal or by medications that slow gastric emptying. Plasma Cmax and AUC of bexagliflozin increase in a dose-proportional manner following single doses from 3 mg (0.15 times the recommended dose) to 90 mg (4.5 times the recommended dose).
Effect of Food
Administration of bexagliflozin after consumption of a standard high-fat, high-caloric meal increased Cmax and AUC by 31% and 10%, respectively, compared to dosing in the fasted state. The median Tmax was increased to 5 hours. The effects of food on bexagliflozin pharmacokinetics are not considered clinically relevant.
Distribution
Bexagliflozin is approximately 93% bound to plasma protein. Neither renal nor hepatic impairment substantially alters protein binding. The apparent volume of distribution is 262 L.
Elimination
Metabolism
Bexagliflozin is mainly metabolized by UGT1A9 and, to a lesser extent, CYP3A. In plasma the most abundant metabolite is the pharmacologically inactive 3′-O-glucuronide, which was found to constitute 32.2% of the parent compound AUC in a radiolabeled tracer study. None of the metabolites are expected to have clinically relevant pharmacological effects.
Excretion
The apparent oral clearance of bexagliflozin is 19.1 L/h by population pharmacokinetic modeling. The apparent terminal elimination half-life of bexagliflozin was approximately 12 hours. Following administration of an oral [14C]-bexagliflozin solution to healthy subjects, 91.6% of input radioactivity was recovered, 51.1% in feces, the majority as bexagliflozin, and 40.5% in urine, largely as the 3′-O-glucuronide. The proportion of input radioactivity recovered as bexagliflozin in urine and feces was 1.5% and 28.7%, respectively
Specific Populations
Patients with Renal Impairment
In a clinical pharmacology study in patients with type 2 diabetes mellitus and mild (eGFR 60 to 89 mL/min/1.73 m²), moderate (eGFR 30 to 59 mL/min/1.73 m²), and severe (eGFR less than 30 mL/min/1.73 m²) renal impairment, the AUC of bexagliflozin was 7%, 34% and 54% greater than in patients with normal renal function, respectively, after administration of a single 20 mg dose of bexagliflozin. These increases in bexagliflozin AUC are not considered clinically meaningful.
Consistent with the mechanism of action of bexagliflozin, the 24-hour UGE in patients with type 2 diabetes mellitus and mild, moderate, and severe renal impairment was 17%, 60%, and 83% lower than in patients with type 2 diabetes mellitus with normal renal function, respectively. Therefore, the glucose-lowering pharmacodynamic response to bexagliflozin declines with increasing severity of renal impairment. The impact of hemodialysis on bexagliflozin exposure is not known.
Patients with Hepatic Impairment
In patients with moderate hepatic impairment (Child-Pugh class B), the AUC of bexagliflozin increased by 28%, and Cmax increased by 6.3% compared to subjects with normal hepatic function. These increases in bexagliflozin AUC and Cmax are not considered clinically meaningful. There is no clinical experience in patients with Child-Pugh class C (severe) hepatic impairment.
Effects of Age, Body Weight, Sex, and Race
Based on a population pharmacokinetic analysis, age, body weight, sex and race do not have a clinically relevant effect on the pharmacokinetics of bexagliflozin.
Drug Interaction Studies
In vitro Assessment of Drug Interactions
Based on in vitro studies, bexagliflozin is not expected to inhibit CYP450 isoenzymes (CYPs) 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, and 3A4, or induce CYPs 1A2, 2C19 and 3A4 at clinically relevant plasma concentrations.
Bexagliflozin is not expected to inhibit drug transporters including breast cancer resistance protein (BRCP), bile salt export pump (BSEP), organic anion transporting polypeptides (OATP1B1, OATP1B3), anion transporters (OAT1, OAT3), organic cation transporters (OCT1, OCT2), and multidrug and toxin extrusion transporters (MATE1, MATE2-K) at clinically relevant plasma concentrations. Bexagliflozin is a substrate for P-glycoprotein (P-gp).
In vivo Assessment of Drug Interactions
There are no clinically meaningful changes in bexagliflozin exposure when taken with metformin, glimepiride, sitagliptin, exenatide, probenecid, or verapamil (Figure 1). Bexagliflozin had no clinically relevant effect on the pharmacokinetics of metformin, glimepiride, sitagliptin, and digoxin (Figure 2).
Figure 1. Effect of Other Drugs on the Pharmacokinetics of Bexagliflozin:
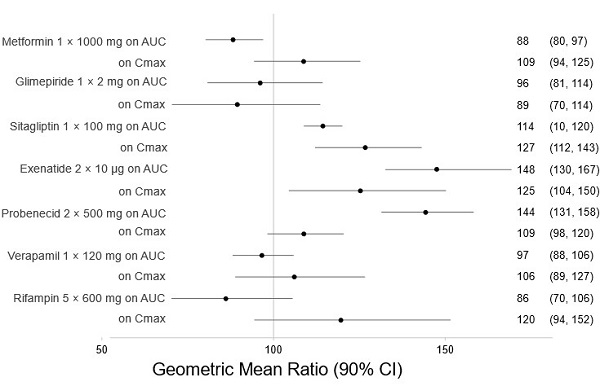
Note: Rifampin dosing over 5 days, the regimen may not represent the maximal impact on bexagliflozin exposure. All others were single day dosing (once daily or twice daily).
Figure 2. Effect of Bexagliflozin on the Pharmacokinetics of Other Drugs
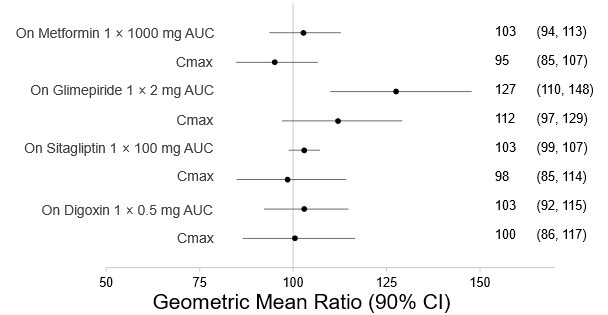
Note: Single day dosing.
Related medicines
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.