Ceritinib
Chemical formula: C₂₈H₃₆ClN₅O₃S Molecular mass: 558.135 g/mol PubChem compound: 57379345
Interactions
Ceritinib interacts in the following cases:
CYP2A6 substrates, CYP2E1 substrates
Based on in vitro data, ceritinib also inhibits CYP2A6 and CYP2E1 at clinically relevant concentrations. Therefore, ceritinib may have the potential to increase plasma concentrations of co-administered medicinal products that are predominantly metabolised by these enzymes. Caution should be exercised with concomitant use of CYP2A6 and CYP2E1 substrates and ADRs carefully monitored.
CYP3A substrates, CYP2C9 substrates
Based on in vitro data, ceritinib competitively inhibits the metabolism of a CYP3A substrate, midazolam, and a CYP2C9 substrate, diclofenac. Time-dependent inhibition of CYP3A was also observed. The steady-state Cmax value of ceritinib at the dose of 450 mg daily taken with food may exceed the Ki values for CYP3A and CYP2C9, suggesting that ceritinib could inhibit the clearance of other medicinal products metabolised by these enzymes at clinically relevant concentrations. Dose reduction may be needed for co-administered medicinal products that are predominantly metabolised by CYP3A and CYP2C9. Co-administration of ceritinib with CYP3A substrates known to have narrow therapeutic indices (e.g. astemizole, cisapride, ciclosporin, ergotamine, fentanyl, pimozide, quinidine, tacrolimus, alfentanil and sirolimus) and CYP2C9 substrates known to have narrow therapeutic indices (e.g. phenytoin and warfarin) should be avoided.
P-gp inhibitors
Based on in vitro data, ceritinib is a substrate of the efflux transporter P-glycoprotein (P-gp). If ceritinib is administered with medicinal products that inhibit P-gp, an increase in ceritinib concentration is likely. Caution should be exercised with concomitant use of P-gp inhibitors and ADRs carefully monitored.
Strong CYP3A inhibitors
Avoid concomitant use of strong CYP3A inhibitors during treatment with ceritinib. If concomitant use of a strong CYP3A inhibitor is unavoidable, reduce the dose by approximately one third (dose not clinically verified), rounded to the nearest multiple of the 150 mg dosage strength. Patients should be carefully monitored for safety.
If long-term concomitant treatment with a strong CYP3A inhibitor is necessary and the patient tolerates the reduced dose well, the dose may be increased again with careful monitoring for safety, to avoid potential under-treatment.
After discontinuation of a strong CYP3A inhibitor, resume at the dose that was taken prior to initiating the strong CYP3A inhibitor.
In healthy subjects, co-administration of a single 450 mg fasted ceritinib dose with ketoconazole (200 mg twice daily for 14 days), a strong CYP3A/P-gp inhibitor, resulted in 2.9-fold and 1.2-fold increase in ceritinib AUCinf and Cmax, respectively, compared to when ceritinib was given alone. The steady-state AUC of ceritinib at reduced doses after co-administration with ketoconazole 200 mg twice daily for 14 days was predicted by simulations to be similar to the steady-state AUC of ceritinib alone. Avoid concomitant use of strong CYP3A inhibitors during treatment with ceritinib. If it is not possible to avoid concomitant use with strong CYP3A inhibitors (including, but not limited to, ritonavir, saquinavir, telithromycin, ketoconazole, itraconazole, voriconazole, posaconazole and nefazodone), reduce the ceritinib dose by approximately one third, rounded to the nearest multiple of the 150 mg dosage strength. After discontinuation of a strong CYP3A inhibitor, resume the ceritinib dose that was taken prior to initiating the strong CYP3A inhibitor.
Strong CYP3A inducers, P-gp inducers
In healthy subjects, co-administration of a single 750 mg fasted ceritinib dose with rifampicin (600 mg daily for 14 days), a strong CYP3A/P-gp inducer, resulted in 70% and 44% decreases in ceritinib AUCinf and Cmax, respectively, compared to when ceritinib was given alone. Co-administration of ceritinib with strong CYP3A/P-gp inducers decreases ceritinib plasma concentrations. Concomitant use of strong CYP3A inducers should be avoided; this includes, but is not limited to, carbamazepine, phenobarbital, phenytoin, rifabutin, rifampicin and St. John's Wort (Hypericum perforatum). Caution should be exercised with concomitant use of P-gp inducers.
Severe renal impairment
A dedicated pharmacokinetic study in patients with renal impairment has not been conducted. However, based on available data, ceritinib elimination via the kidney is negligible. Therefore, no dose adjustment is necessary in patients with mild to moderate renal impairment. Caution should be used in patients with severe renal impairment, as there is no experience with ceritinib in this population.
Severe hepatic impairment
Based on available data, ceritinib is eliminated primarily via the liver. Particular caution should be exercised when treating patients with severe hepatic impairment and the dose should be reduced by approximately one third, rounded to the nearest multiple of the 150 mg dosage strength.
Proton pump inhibitors, H2-receptor antagonists, antacids
Ceritinib demonstrates pH-dependent solubility and becomes poorly soluble as pH increases in vitro. Acid reducing agents (e.g. proton pump inhibitors, H2-receptor antagonists, antacids) can alter the solubility of ceritinib and reduce its bioavailability. Co-administration of a single 750 mg fasted ceritinib dose with a proton pump inhibitor (esomeprazole) 40 mg daily for 6 days in healthy, fasting subjects decreased ceritinib AUC by 76% and Cmax by 79%.
The drug-drug interaction study was designed to observe the impact of proton pump inhibitor in the worst scenario, but in clinical use the impact of proton pump inhibitor on ceritinib exposure appears to be less pronounced. A dedicated study to evaluate the effect of gastric acid-reducing agents on the bioavailability of ceritinib under steady state has not been conducted. Caution is advised with concomitant use of proton pump inhibitors, as exposure of ceritinib may be reduced.
There is no data with concomitant use of H2 blockers or antacids. However, the risk for a clinically relevant decrease in bioavailability of ceritinib is possibly lower with concomitant use of H2 blockers if they are administered 10 hours before or 2 hours after the ceritinib dose, and with antacids if they are administered 2 hours before or 2 hours after the ceritinib dose.
Oral contraceptives
The effectiveness of concomitant administration of oral contraceptives may be reduced.
QT prolongation, medicinal products that may lead to QT prolongation
In clinical studies, QT prolongation was observed with ceritinib. Therefore, ceritinib should be used with caution in patients who have or may develop prolongation of the QT interval, including those patients taking anti-arrhythmic medicinal products such as class I (e.g. quinidine, procainamide, disopyramide) or class III (e.g. amiodarone, sotalol, dofetilide, ibutilide) anti-arrhythmics or other medicinal products that may lead to QT prolongation such as astemizole, domperidone, droperidol, chloroquine, halofantrine, clarithromycin, haloperidol, methadone, cisapride and moxifloxacin. Monitoring of the QT interval is indicated in the event of combinations of such medicinal products.
Use of ceritinib in patients with congenital long QT syndrome should be avoided. The benefits and potential risks of ceritinib should be considered before beginning therapy in patients who have pre-existing bradycardia (heart rate less than 60 beats per minute [bpm]), patients who have a history of or predisposition for QTc prolongation, patients who are taking anti-arrhythmics or other medicinal products that are known to prolong the QT interval and patients with relevant pre-existing cardiac disease and/or electrolyte disturbances. Periodic monitoring with ECGs and periodic monitoring of electrolytes (e.g. potassium) is recommended in these patients. In the event of vomiting, diarrhoea, dehydration or impaired renal function, correct electrolytes as clinically indicated. Ceritinib should be permanently discontinued in patients who develop QTc >500 msec or >60 msec change from baseline and torsade de pointes or polymorphic ventricular tachycardia or signs/symptoms of serious arrhythmia. Ceritinib should be withheld in patients who develop QTc >500 msec on at least two separate ECGs until recovery to baseline or a QTc ≤480 msec, then reinitiated with dose reduced by 150 mg.
Interstitial lung disease, pneumonitis
Severe, life-threatening or fatal ILD/pneumonitis have been observed in patients treated with ceritinib in clinical studies. Most of these severe/life-threatening cases improved or resolved with interruption of treatment.
Patients should be monitored for pulmonary symptoms indicative of ILD/pneumonitis. Other potential causes of ILD/pneumonitis should be excluded, and ceritinib permanently discontinued in patients diagnosed with any grade treatment-related ILD/pneumonitis.
Hepatotoxicity
Cases of hepatotoxicity occurred in 1.1% of patients receiving ceritinib in clinical studies. Increases to grade 3 or 4 ALT elevations were observed in 25% of patients. The majority of cases were manageable with dose interruption and/or dose reduction. Few events required discontinuation of treatment.
Patients should be monitored with liver laboratory tests (including ALT, AST and total bilirubin) prior to the start of treatment, every 2 weeks during the first three months of treatment and monthly thereafter. In patients who develop transaminase elevations, more frequent monitoring of liver transaminases and total bilirubin should be carried out as clinically indicated. Particular caution should be exercised when treating patients with severe hepatic impairment, and the dose should be adjusted. Limited experience in these patients showed a worsening of the underlying condition (hepatic encephalopathy) in 2 out of 10 patients exposed to 750 mg single doses of ceritinib under fasted conditions. Other factors apart from study treatment could have impacted on observed events of hepatic encephalopathy, however, the relation between study treatment and events cannot be fully ruled out. No dose adjustment is necessary in patients with mild or moderate hepatic impairment.
Diarrhoea, nausea, vomiting
Diarrhoea, nausea, or vomiting occurred in 74.2% of 89 patients treated with ceritinib at the
recommended dose of 450 mg taken with food in a dose optimisation study and were mainly grade 1 events (49.4%). One patient (1.1%) experienced grade 3 diarrhoea. Seven patients (7.9%) required study drug interruption due to diarrhoea or nausea. The incidence and severity of gastrointestinal adverse drug reactions were higher for patients treated with ceritinib 750 mg fasted (diarrhoea 76%, nausea 50%, vomiting 56%; 12% reported a grade 3/4 event) compared to 450 mg with food (diarrhoea 56%, nausea 45%, vomiting 35%; 1.1% reported a grade 3/4 event).
No patients required dose reduction or discontinuation of ceritinib due to diarrhoea, nausea or vomiting.
Patients should be monitored and managed using standards of care, including anti-diarrhoeals, anti-emetics or fluid replacement, as clinically indicated. Dose interruption and dose reduction should be employed as necessary. If vomiting occurs during the course of treatment, the patient should not take an additional dose, but should continue with the next scheduled dose.
Hyperglycaemia
Cases of hyperglycaemia (all grades) have been reported in less than 10% of patients treated with ceritinib in clinical studies; grade 3-4 hyperglycaemia was reported in 5.4% of patients. The risk of hyperglycaemia was higher in patients with diabetes mellitus and/or concurrent steroid use.
Patients should be monitored for fasting plasma glucose prior to the start of ceritinib treatment and periodically thereafter as clinically indicated. Anti-hyperglycaemic medicinal products should be initiated or optimised as indicated.
Bradycardia, beta blockers, non-dihydropyridine calcium channel blockers, clonidine, digoxin
Asymptomatic cases of bradycardia (heart rate less than 60 bpm) have been observed in 21 out of 925 (2.3%) patients treated with ceritinib in clinical studies.
Use of ceritinib in combination with other agents known to cause bradycardia (e.g. beta blockers, non-dihydropyridine calcium channel blockers, clonidine and digoxin) should be avoided as far as possible. Heart rate and blood pressure should be monitored regularly. In cases of symptomatic bradycardia that is not life-threatening, ceritinib should be withheld until recovery to asymptomatic bradycardia or to a heart rate of 60 bpm or above, the use of concomitant medicinal products should be evaluated and the ceritinib dose adjusted if necessary. In the event of life-threatening bradycardia ceritinib should be permanently discontinued if no contributing concomitant medicinal product is identified; however, if associated with a concomitant medicinal product known to cause bradycardia or hypotension, ceritinib should be withheld until recovery to asymptomatic bradycardia or to a heart rate of 60 bpm or above. If the concomitant medicinal product can be adjusted or discontinued, ceritinib should be reinitiated with dose reduced by 150 mg on recovery to asymptomatic bradycardia or to a heart rate of 60 bpm or above, with frequent monitoring.
Pregnancy
There are no or limited amount of data from the use of ceritinib in pregnant women.
Animal studies are insufficient with respect to reproductive toxicity.
Ceritinib should not be used during pregnancy unless the clinical condition of the woman requires treatment with ceritinib.
Nursing mothers
It is unknown whether ceritinib/metabolites are excreted in human milk. A risk to the newborn/infant cannot be excluded.
A decision must be made whether to discontinue breast-feeding or discontinue/abstain from ceritinib therapy taking into account the benefit of breast-feeding for the child and the benefit of therapy for the woman.
Carcinogenesis, mutagenesis and fertility
Women of childbearing potential
Women of childbearing potential should be advised to use a highly effective method of contraception while taking ceritinib and for up to 3 months after discontinuing treatment.
Fertility
The potential for ceritinib to cause infertility in male and female patients is unknown.
Effects on ability to drive and use machines
Ceritinib has minor influence on the ability to drive or use machines. Caution should be exercised when driving or using machines during treatment as patients may experience fatigue or vision disorders.
Adverse reactions
Summary of the safety profile
Adverse drug reactions (ADRs) described below reflect exposure to ceritinib 750 mg once daily fasted in 925 patients with ALK-positive advanced NSCLC across a pool of seven clinical studies including two randomised, active-controlled, phase 3 studies (studies A2301 and A2303).
The median duration of exposure to ceritinib 750 mg fasted was 44.9 weeks (range: 0.1 to 200.1 weeks).
ADRs with an incidence of ≥10% in patients treated with ceritinib 750 mg fasted were diarrhoea, nausea, vomiting, fatigue, liver laboratory test abnormalities, abdominal pain, decreased appetite, weight decreased, constipation, blood creatinine increased, rash, anaemia and oesophageal disorder.
Grade 3-4 ADRs with an incidence of ≥5% in patients treated with ceritinib 750 mg fasted were liver laboratory test abnormalities, fatigue, vomiting, hyperglycaemia, nausea and diarrhoea.
In the dose optimisation study A2112 (ASCEND-8) in both previously treated and untreated patients with ALK-positive advanced NSCLC, the overall safety profile of ceritinib at the recommended dose of 450 mg with food (N=89) was consistent with ceritinib 750 mg fasted (N=90), except for a reduction in gastrointestinal adverse drug reactions, while achieving comparable steady-state exposure.
Tabulated list of ADRs
Table 2 shows the frequency category of ADRs reported for ceritinib in patients treated at a dose of 750 mg fasted (N=925) in seven clinical studies. The frequency of selected gastrointestinal ADRs (diarrhoea, nausea and vomiting) are based on patients treated with a dose of 450 mg once-daily with food (N=89).
ADRs are listed according to MedDRA system organ class. Within each system organ class, the ADRs are ranked by frequency, with the most frequent reactions first. In addition, the corresponding frequency category using the following convention (CIOMS III) is also provided for each ADR: very common (≥1/10); common (≥1/100 to <1/10); uncommon (≥1/1,000 to <1/100); rare (≥1/10,000 to <1/1,000); very rare (<1/10,000); and not known (cannot be estimated from the available data).
Table 2. ADRs in patients treated with ceritinib:
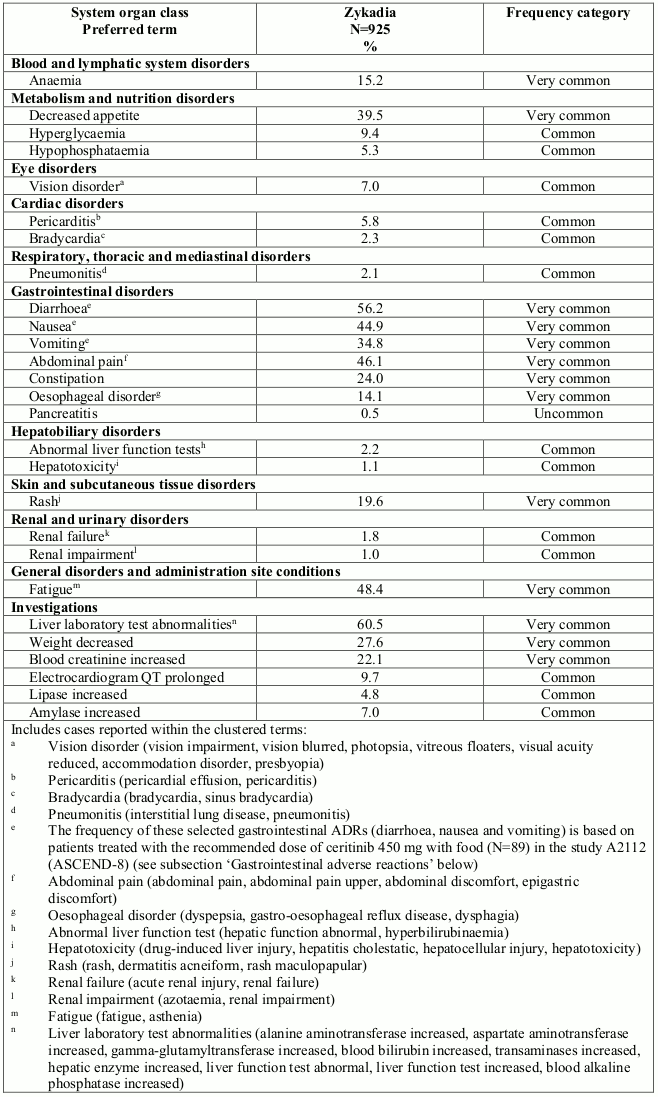
Elderly (≥65 years)
Across seven clinical studies, 168 out of 925 patients (18.2%) treated with ceritinib were aged 65 years or older. The safety profile in patients aged 65 years or older was similar to that in patients less than 65 years of age. There are no safety data in patients older than 85 years of age.
Hepatotoxicity
Concurrent elevations of ALT or AST greater than 3× ULN and total bilirubin greater than 2× ULN without elevated alkaline phosphatase have been observed in less than 1% of patients in clinical studies with ceritinib. Increases to grade 3 or 4 ALT elevations were observed in 25% of patients receiving ceritinib. Hepatotoxicity events were managed with dose interruptions or reductions in 40.6% of patients. 1% of patients required permanent discontinuation of treatment in clinical studies with ceritinib.
Liver laboratory tests including ALT, AST and total bilirubin should be performed prior to the start of treatment, every 2 weeks during the first three months of treatment and monthly thereafter, with more frequent testing for grade 2, 3 or 4 elevations. Patients should be monitored for liver laboratory test abnormalities and managed.
Gastrointestinal adverse reactions
Nausea, diarrhoea and vomiting were among the most commonly reported gastrointestinal events. In the dose optimisation study A2112 (ASCEND-8) in both previously treated and untreated patients with ALK-positive advanced NSCLC at the recommended dose of ceritinib 450 mg taken with food (N=89), adverse events of diarrhoea, nausea and vomiting were mainly grade 1 (49.4%). A grade 3 event of diarrhoea was reported in one patient (1.1%). Gastrointestinal events were managed primarily with concomitant medicinal products including anti-emetic/anti-diarrhoeal medicinal products. Seven patients (7.9%) required study drug interruption due to diarrhoea or nausea. No patients had diarrhoea, nausea, or vomiting that required dose reduction or discontinuation of study drug. The incidence and severity of gastrointestinal adverse drug reactions were reduced for patients treated with ceritinib 450 mg with food (diarrhoea 56%, nausea 45%, vomiting 35%; 1.1% reported a grade ¾ event) compared to 750 mg fasted (diarrhoea 76%, nausea 50%, vomiting 56%; 12% reported a grade ¾ event). Patients should be managed.
QT interval prolongation
QTc prolongation has been observed in patients treated with ceritinib. Across the seven clinical studies, 9.7% of patients treated with ceritinib had events of QT prolongation (any grade), including grade 3 or 4 events in 2.1% of patients. These events required dose reduction or interruption in 2.1% of patients and led to discontinuation in 0.2% of patients.
Treatment with ceritinib is not recommended in patients who have congenital long QT syndrome or who are taking medicinal products known to prolong the QTc interval. Particular care should be exercised when administering ceritinib to patients with an increased risk of experiencing torsade de pointes during treatment with a QTc-prolonging medicinal product.
Patients should be monitored for QT prolongation and managed.
Bradycardia
Across the seven clinical studies, bradycardia and/or sinus bradycardia (heart rate less than 60 bpm) events (all grade 1) were reported in 2.3% of patients. These events required dose reduction or interruption in 0.2% of patients. None of these events led to discontinuation of ceritinib treatment. The use of concomitant medicinal products associated with bradycardia should be carefully evaluated. Patients who develop symptomatic bradycardia should be managed.
Interstitial lung disease/Pneumonitis
Severe, life-threatening, or fatal interstitial lung disease (ILD)/pneumonitis have been observed in patients treated with ceritinib. Across the seven clinical studies, any grade ILD/pneumonitis has been reported in 2.1% of patients treated with ceritinib, and grade 3 or 4 events have been reported in 1.2% of patients. These events required dose reduction or interruption in 1.1% of patients and led to discontinuation in 0.9% of patients. Patients with pulmonary symptoms indicative of ILD/pneumonitis should be monitored. Other potential causes of ILD/pneumonitis should be excluded.
Hyperglycaemia
Hyperglycaemia (all grades) was reported in 9.4% of patients treated with ceritinib across the seven clinical studies; grade 3 or 4 events were reported in 5.4% of patients. These events required dose reduction or interruption in 1.4% of patients and led to discontinuation in 0.1% of patients. The risk of hyperglycaemia was higher in patients with diabetes mellitus and/or concurrent steroid use. Monitoring of fasting serum glucose is required prior to the start of ceritinib treatment and periodically thereafter as clinically indicated. Administration of anti-hyperglycaemic medicinal products should be initiated or optimised as indicated.
© All content on this website, including data entry, data processing, decision support tools, "RxReasoner" logo and graphics, is the intellectual property of RxReasoner and is protected by copyright laws. Unauthorized reproduction or distribution of any part of this content without explicit written permission from RxReasoner is strictly prohibited. Any third-party content used on this site is acknowledged and utilized under fair use principles.